In the context of their close resemblance to natural enzymatic systems, tetrapyrrole metal complexes, such as heme, chlorophyll, vitamin B12, and coenzyme F430, have attracted considerable attention and are among the most extensively studied subjects in bio-related coordination chemistry [[1], [2], [3]]. In addition to the traditional tetrapyrrolic frameworks, many synthetic porphyrin analogs (porphyrinoids) have also been explored for their metal-binding capabilities [[4], [5], [6]]. These exotic metallo-porphyrinoids exhibit structure-dependent chemical and physical properties, including their applicability in photoacoustic (PA) imaging, enhancement of photodynamic therapy (PDT), fluorescence tagging, light-harvesting, electron transfer, catalytic transformations, and sensing capabilities, all of which hold great promise for various material applications [[7], [8], [9], [10]].
Regarding the coordination behavior of porphyrin molecules in solution, it has traditionally been understood as follows: deformation of the porphyrin macrocycle, coordination of solvated metal ions to the porphyrin ring, deprotonation of a pyrrolic hydrogen atom, and subsequent bonding between a nitrogen site and the metal ion [11,12]. The intermediate species, characterized by a doubly protonated porphyrin with a metal ion at the central NNNN core, is recognized as a “sitting-atop” complex [13]. Recently, researchers reported a novel class of “sitting-atop” semi-sandwich complexes derived from metal-free porphycenes, where the ruthenium(II) pentamethylcyclopentadienyl ([RuCp*]+) fragment resides within the NNNN core. In this case, the metal ion is situated on a distinct π-electron surface of the porphyrin plane, forming sandwich metal complexes [[14], [15], [16], [17]]. Additionally, unique cis-bimetallic Pd(II) complexation within the NNNN site of the macrocycles with acetylacetonate ligands has been reported for rigidified porphycene derivatives [18].
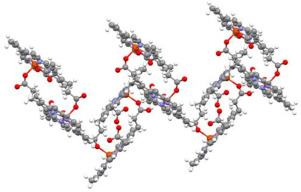
Based on these insights, out-of-plane metal complexation of porphyrin macrocycles allows the formation of semi-sandwich and full-sandwich species, enabling modulation of the inherent optical and electronic properties of specific metalloporphyrins. Ligands with highly planar delocalized π-electron systems facilitate the synthesis of sandwich-type complexes. For example, ferrocene (Fc) represents a classic sandwich metal complex comprising two 6π-electron aromatic cyclopentadienyl (Cp) ligands and a d6 iron(II) center [19,20]. Consequently, the quest for novel sandwich complexes has remained a fundamental pursuit in organometallic chemistry, with isolated bis(cyclopentadienyl) transition metal complexes of ruthenium, cobalt, nickel, rhodium, iridium, and others having been reported to date [[21], [22], [23], [24]]. Similarly, light-responsive reactions have been effectively demonstrated in binuclear complexes through ligand modification. For instance, chloride-bridged binuclear complexes of the general formula [(π-ligand)M-μ-Cl2]2 represent a widely used class of efficient photoactive precursors (e.g., [CpRh(μ-Cl)2]2, [(cod)RhCl]2) (cod = cyclooctadiene) (Fig. 1a). In [CpRh(μ-Cl)2]2, bond cleavage occurs at the weakly coordinating Rh–Cl bridging bond, generating a reactive, unsaturated Cp*‑rhodium fragment, which subsequently participates in bond-breaking and formation processes [[25], [26], [27], [28]]. However, binuclear metal complexes, known for their distinctive optical, redox, and electronic properties, have not been as extensively investigated as their mononuclear counterparts.
Considering the intriguing functionalities of porphyrins, we have explored a unique analog known as N-fused porphyrin (1; NFP), which can be viewed as isoelectronic to monoanionic cyclopentadienyl (Cp) and trispyrazolylborate (Tp). The NFP serves as an ideal ligand platform for metal complexation due to its monoanionic, tridentate N3 donor, organized in a fac manner [29,30]. The inner-fused chromophore, formed by inverting the N-confused pyrrole ring and subsequently forming an oxidative N–C bond with the adjacent pyrrole nitrogen atom of the N-confused porphyrin (NCP), results in a unique inner-fused [5.5.5]-tri-pentacyclic ring with an 18π-aromatic circuit. Notably, the π-conjugated structure of NFP leads to longer wavelength absorption, extending up to 1000 nm in the near-infrared (NIR) region [31,32]. Symmetry plays a crucial role in determining the distinctive structural and photophysical properties of NFP. Introducing the N-fusion domain leads to electronic perturbations, resulting in asymmetric Cs-type structures. This reduction in symmetry breaks degeneracy, causing the splitting of Bx and By as well as Qx and Qy transitions. Consequently, it impacts the Soret and Q bands, leading to optical features such as the emergence of a NIR band with relatively higher intensity [33,34]. Moreover, the NFP precursor, 21-bromo-N-fused porphyrin (1-Br), can extend π-conjugation within the N-fused macrocycle through aryl-ethynyl substitutions [35,36].
Of paramount importance is the direct correlation between the isoelectronic behavior of 1 and Cp in the synthesis of NFP metallocenes, including (i) the NFp double-decker ferrocene-like complex, FeII(NFp)2 and (ii) the heteroleptic ruthenocene sandwich complex ligated by the aromatic NFP ring and Cp, [RuII(Cp)(NFp)] [37,38] (Fig. 1b). Additionally, various half-sandwich complexes involving ruthenium, rhenium, manganese, tungsten, rhodium, and iridium have been observed, featuring oxygen, carbonyl, chlorides, cyclooctadiene (cod), etc., that stabilize the NFp-metal coordination environment [5,32].
Recent investigations have revealed that when the redox-active NFP chromophore coordinates with catalytically active metals like rhodium or iridium, dynamic cyclooctadiene isomerization and aromatic ring cleavage occur, along with cyclooctadiene isomerization [39,40]. Therefore, the distinctive NFp‑rhodium chemistry can be further harnessed to explore a wide range of binuclear complexes, drawing inspiration from the well-known Cp*-based organometallic reagent, pentamethylcyclopentadienyl rhodium(III) dichloride dimer, [Cp*RhCl2]2, in this study. Moreover, replacing Cp with a NIR-active chromophore 1 can generate an entirely new class of π-extended analogs. These photoactive analogs could prove instrumental in exploring applications arising from photothermal processes.



留言 (0)