
記住我

Systemic lupus erythematosus (SLE) is an autoimmune disease that affects multiple systems and can occasionally manifest as hematological disorders and gastrointestinal (GI) tract abnormalities.1 Lupus enteritis (LE), a rare complication of SLE resulting from inflammation or vasculitis of the small bowel, presents with nonspecific symptoms, primarily abdominal pain, vomiting, and diarrhea.2 Though likely underdiagnosed, an estimated 0.2%–5.8% of patients with SLE develop LE.3 The diagnosis of LE is clinical and usually involves extensive serological testing and imaging. Serological abnormalities may include leukopenia, anemia, hypocomplementemia, and positive SLE antibodies.1,4,5 Computed tomography scan can aid in the diagnosis of LE, often showing bowel wall thickening, mesenteric fat attenuation, and mesenteric vasodilation.4 Endoscopy is generally nondiagnostic but has utility in diagnosing pathologies with overlapping symptoms, such as inflammatory bowel disease.3,4 Treatment is tailored to disease severity, and although most patients are successfully treated with glucocorticoid therapy and have an excellent prognosis, immunosuppression and occasionally intravenous immunoglobulin and plasmapheresis are used in severe cases.2,4,6,7 We describe a unique case of a patient with SLE complicated by severe LE, intractable GI bleeding, disseminated intravascular coagulation (DIC), and warm autoimmune hemolytic anemia (WAHA). To the knowledge of the authors, this is the first case reported of LE complicated by intractable GI bleeding with WAHA.
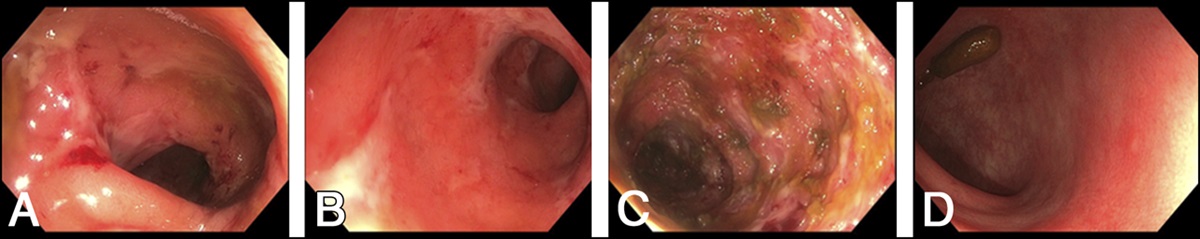
CASE REPORTA 22-year-old woman with Sjögren syndrome and a 1-year history of SLE presented to a tertiary care center with 1 day of nausea, vomiting, nonbloody diarrhea, and severe abdominal pain. She was hospitalized, and initial laboratory findings were indicative of an active SLE flare with elevated anti-double-stranded DNA, increased erythrocyte sedimentation rate, and hypocomplementemia. The patient's initial hemoglobin level was 9.2 g/dL from a baseline of 12.7 g/dL. Extensive infectious workup was negative. Initial abdominal and pelvic computed tomography revealed circumferential wall thickening of the distal small bowel with inflammatory changes and fluid concerning for LE. Inpatient gastroenterology was consulted, and colonoscopy revealed a normal terminal ileum and one transverse colonic ulcer (Figure 1) with reactive colonic mucosa, focal regenerative mucosal change, and lamina propria fibrosis. LE was diagnosed early in the hospital course, and intravenous corticosteroids were administered from day 1 to day 5 of hospitalization. The patient had minimal improvement on methylprednisolone 1 g daily. Repeat abdominal and pelvic computed tomography on day 7 of hospitalization revealed edema and thickening of the stomach, small bowel, and colon (Figure 2). She then developed massive volume hematochezia and a hemoglobin drop to 3.1 g/dL. She underwent repeat colonoscopy on day 8 of hospitalization with similar findings of scattered ulcerations and diffuse oozing not amenable to intervention. Numerous computed tomography angiograms were performed during the hospitalization revealing active bleeding from branches of the superior mesenteric artery, and coil embolization was performed on day 11 of hospitalization. On day 12, a colonoscopy with ileum intubation revealed mid-distal ileal ulcers with punctate hemorrhagic lesions (Figure 3), a large mid-distal ileal ulcer (Figure 4), and a large cecal/ileocecal valve submucosal hematoma treated with electrocautery and hemostatic spray. Biopsies showed mild toxic ischemic-type injury of the ileal mucosa. She was also found to have DIC and WAHA, confirmed with a positive direct antiglobulin test. Throughout the patient's 8-week hospitalization, she received a total of 43 units of packed red blood cells with a nadir hemoglobin of 2.2 g/dL on day 11 (Figure 5). Her hospital stay was also complicated by posterior reversible encephalopathy syndrome, lupus nephritis, cellulitis, acute hypoxic respiratory failure, and critical illness myopathy. The patient was aggressively treated with intravenous methylprednisolone (between day 1 and day 5 of hospitalization), intravenous immunoglobulins (between days 11 and 14), and 2 doses of cyclophosphamide (on day 13 and day 44) and required 5 therapeutic plasma exchanges (between days 15 and 23). She was discharged on slow-taper prednisone, hydroxychloroquine, and cyclophosphamide. The patient achieved lasting clinical and laboratory remission of her symptoms with stable hemoglobin levels up to 1 year after hospitalization.
 Figure 1.:
Figure 1.: Single transverse colon ulcer seen on endoscopic imaging. Arrow pointing to transverse colon ulceration.
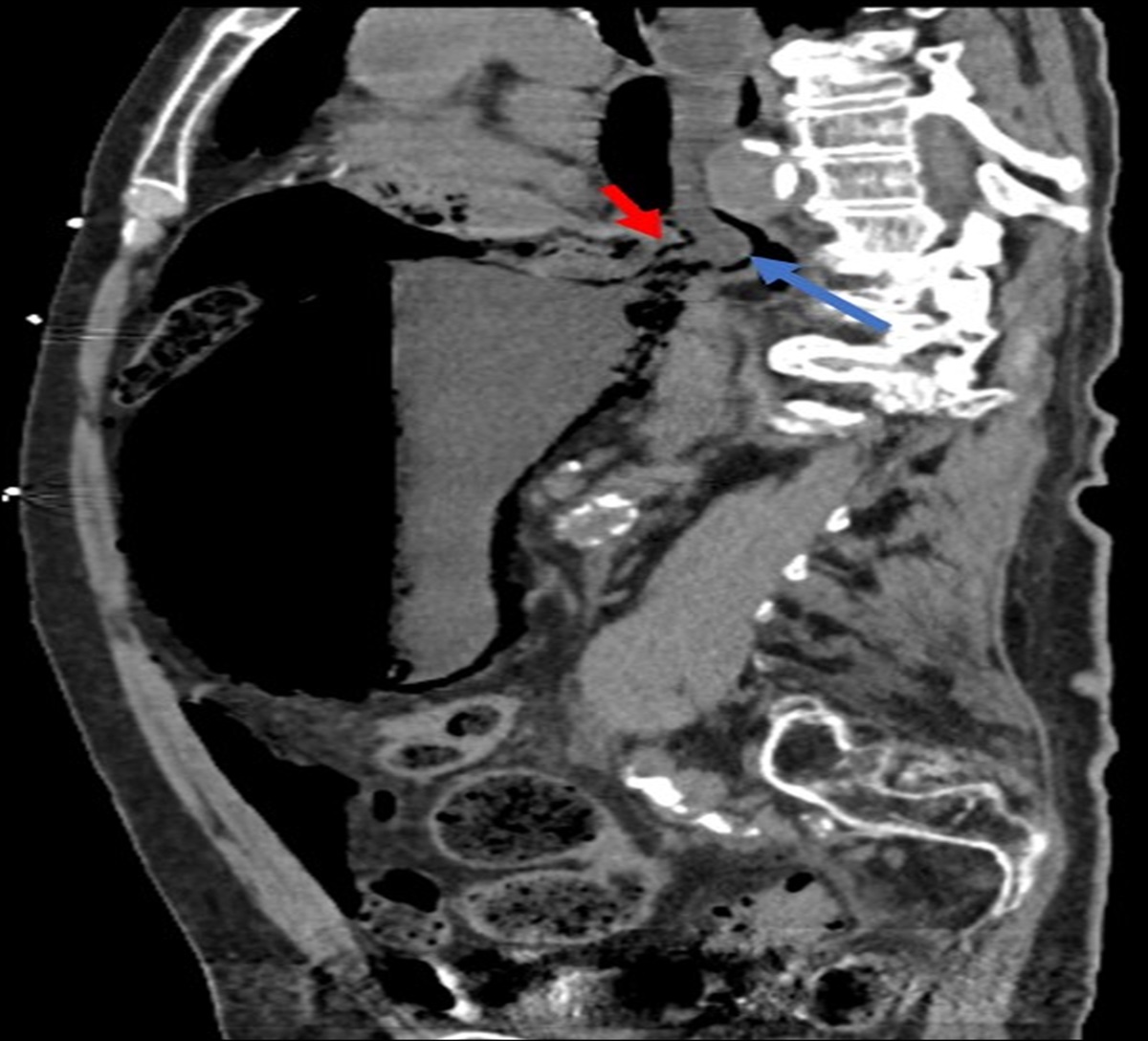
 Figure 2.:
Figure 2.: Abdominal and pelvic computed tomography showing edema and circumferential wall thickening of the stomach, small bowel, and colon characteristic in diagnosis of lupus enteritis.
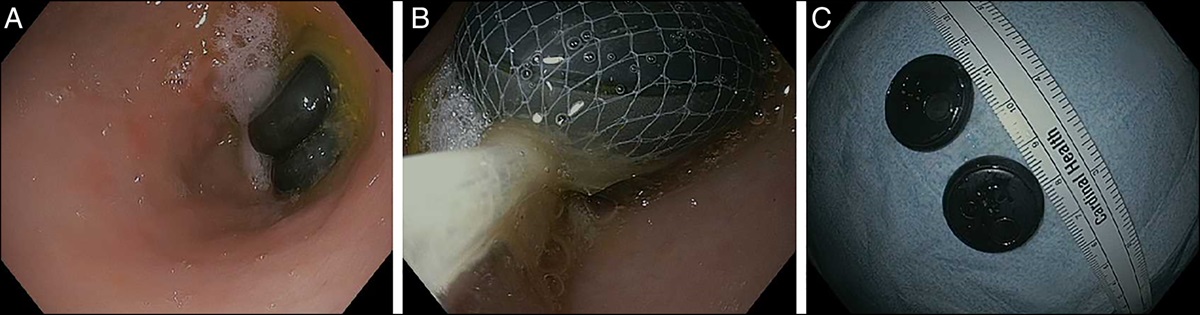
 Figure 3.:
Figure 3.: Mid-distal ileal ulcers with punctate hemorrhagic lesions on endoscopic imaging.
 Figure 4.:
Figure 4.: Single large mid-distal ileal ulcer seen on endoscopic imaging.
 Figure 5.:
Figure 5.: Graph depicting the patient's average Hgb level by day and trends throughout hospitalization countered by the number of blood products transfused daily. The patient's average Hgb level stabilized after the initiation of IVIG and plasmapheresis to a level of approximately 7–9 g/dL. Hgb, hemoglobin; IVIG, intravenous immunoglobulin.
DISCUSSIONLE is a rare but potentially life-threatening complication of SLE, and although most commonly associated with abdominal pain, LE may present with GI bleeding in approximately 3% of cases or even progress to bowel perforation.2,8 In this case of LE manifesting with refractory GI bleeding and acute, multifactorial anemia, the patient demonstrated a lack of improvement despite multiple endoscopic interventions. Endoscopies revealed pancolonic ulcers and distal small bowel wall thickening and inflammation. While ulceration may be present in LE, there does not seem to be consensus on its typical endoscopic presentation. One retrospective study found that over half of patients with LE had normal endoscopic findings.4 Another study found that 60% of its patients who underwent endoscopy had normal macroscopic findings.8 Although endoscopy is often used for thorough evaluation and is helpful to perform interventions on bleeding stigmata, it is difficult to achieve hemostasis in bleeding secondary to autoimmune etiologies.3 In addition, in critically ill patients, repeat endoscopies increase the risk of procedural and sedation-related adverse events. Therefore, we propose that early treatment of the underlying autoimmune disease, in this case with immunosuppression, is the most important factor in achieving long-term hemostasis.
There are currently no available standardized guidelines for the treatment of LE, but most patients with LE experience remission with high-dose corticosteroids with or without immunosuppression.4 The addition of cyclophosphamide immunosuppression has been shown to improve patient prognosis.4 Unfortunately, the patient did not respond to these measures, and her condition was exacerbated by DIC and WAHA, resulting in numerous blood transfusions. Depending on comorbidities, an early multispecialty approach to treatment may facilitate favorable outcomes, ensuring repression of multiorgan involvement and decreasing the likelihood of secondary complications. In this case, early specialist intervention and a multidisciplinary approach including rheumatology, gastroenterology, and nephrology were instrumental in achieving patient remission. Aggressive measures including plasmapheresis and intravenous immunoglobulins led to sustained remission, improvement in the patient's GI symptoms due to LE, and stability in her hemoglobin levels. This case demonstrates the role of early consideration of plasmapheresis in LE in patients experiencing massive, life-threatening GI hemorrhage refractory to standard immunosuppressive therapy.
DISCLOSURESAuthor contributions: A. Thomas and C. Smithhart are the co-first authors of this article and contributed toward data collection and manuscript writing. M. Chopra and A. Jayan contributed toward data collection, manuscript writing and editing, and direct patient care. F. Ali and K. Doughem contributed toward manuscript editing and direct patient care. S. Larson, A. Bhatt and J. Scoon are the senior authors of the manuscript, ensured preservation of data integrity, were in charge of the overall direction and planning of the case report, critically revised the manuscript formation, and contributed toward direct patient care. S. Larson is the article guarantor.
Financial disclosure: None to report.
Previous presentation: This case report was previously presented at ACG Annual Scientific Meeting 2022, October 25, 2022, Charlotte, North Carolina.
Informed consent was obtained for this case report.
REFERENCES 1. Yu C, Gershwin ME, Chang C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J Autoimmun. 2014;48–49:10–3. 2. Sran S, Sran M, Patel N, et al. Lupus enteritis as an initial presentation of systemic lupus erythematosus. Case Rep Gastrointest Med. 2014;2014:962735. 3. Brewer B, Kamen D. Gastrointestinal and hepatic disease in systemic lupus erythematosus. Rheum Dis Clin North Am. 2018;44(1):165–75. 4. Chen L, He Q, Luo M, et al. Clinical features of lupus enteritis: A single-center retrospective study. Orphanet J Rare Dis. 2021;16(1):396. 5. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. 2019;393(10188):2344–58. 6. Leone P, Prete M, Malerba E, et al. Lupus vasculitis: An overview. Biomedicines. 2021;9(11):1626. 7. González Padilla KA, Galarza Delgado DÁ, Garza Elizondo MA, et al. Target sign in woman with abdominal pain and systemic lupus erythematosus treated with plasma exchange. Reumatol Clin. 2016;12(6):348–50. 8. Janssens P, Arnaud L, Galicier L, et al. Lupus enteritis: From clinical findings to therapeutic management. Orphanet J Rare Dis. 2013;8:67.
留言 (0)