
Remember me


Six- to eight-week-old female C57BL/6 mice (20–25 g) were purchased from the Experimental Animal Center of Wuhan University (Wuhan, Hubei, China). Mice were housed in a specific-pathogen-free isolation environment in the Animal Experimental Center of the Renmin Hospital of Wuhan University. Standard laboratory conditions include a controlled temperature (23 ± 1 °C), a 12-h light/dark cycle, a moderate humidity (50–60%) and free access to water and food. All animals were acclimated for 1 week prior to the experiment. Tet2-deficient B6(Cg)-Tet2tm1.2Rao/J mice were obtained from The Jackson Laboratory (West Grove, PA, USA). Wild type (WT) controls and homozygous (Tet2 knockout [Tet2−/−]) mice were bred from heterozygous parents and all mice were genetically identified as described previously (Tan et al. 2022).
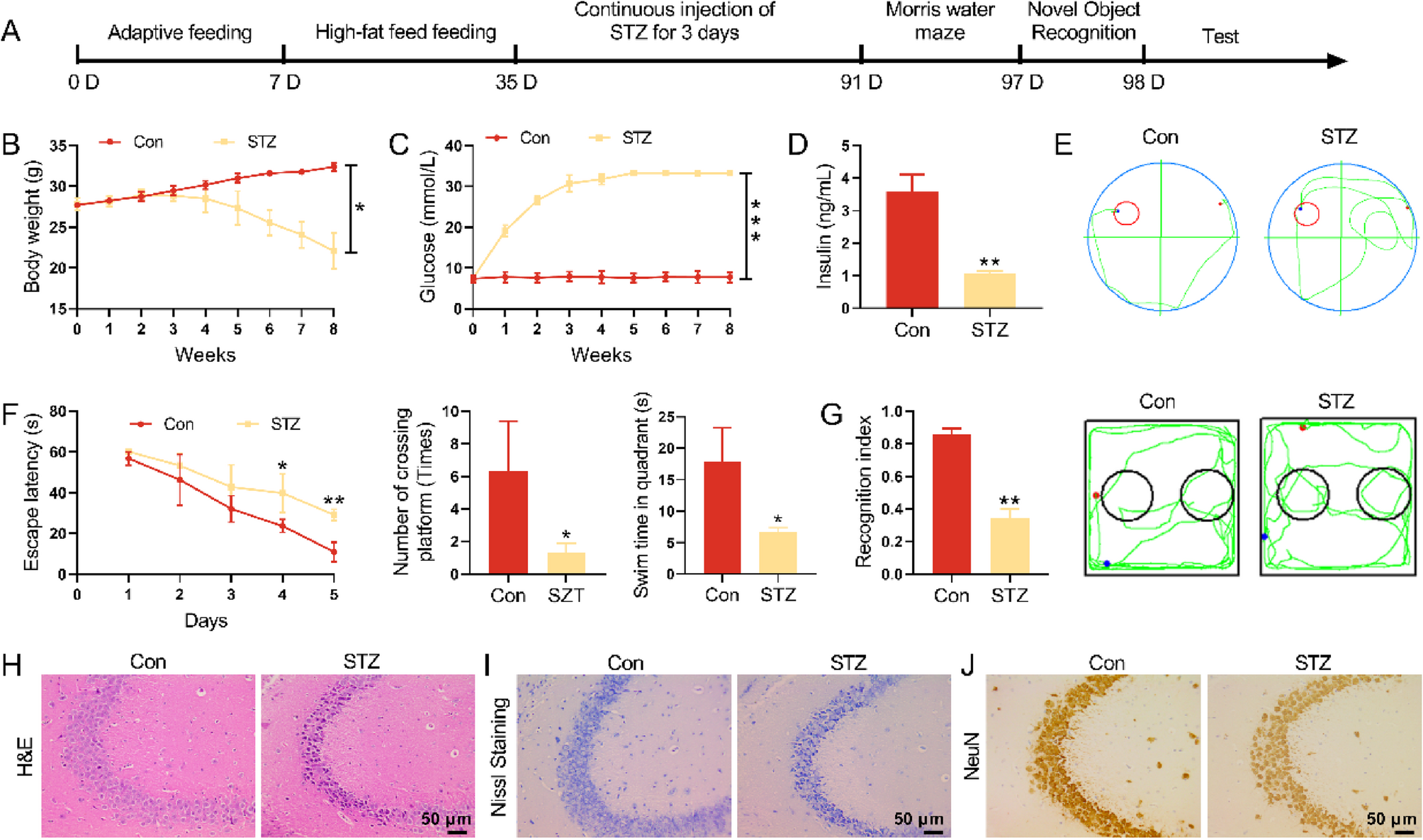
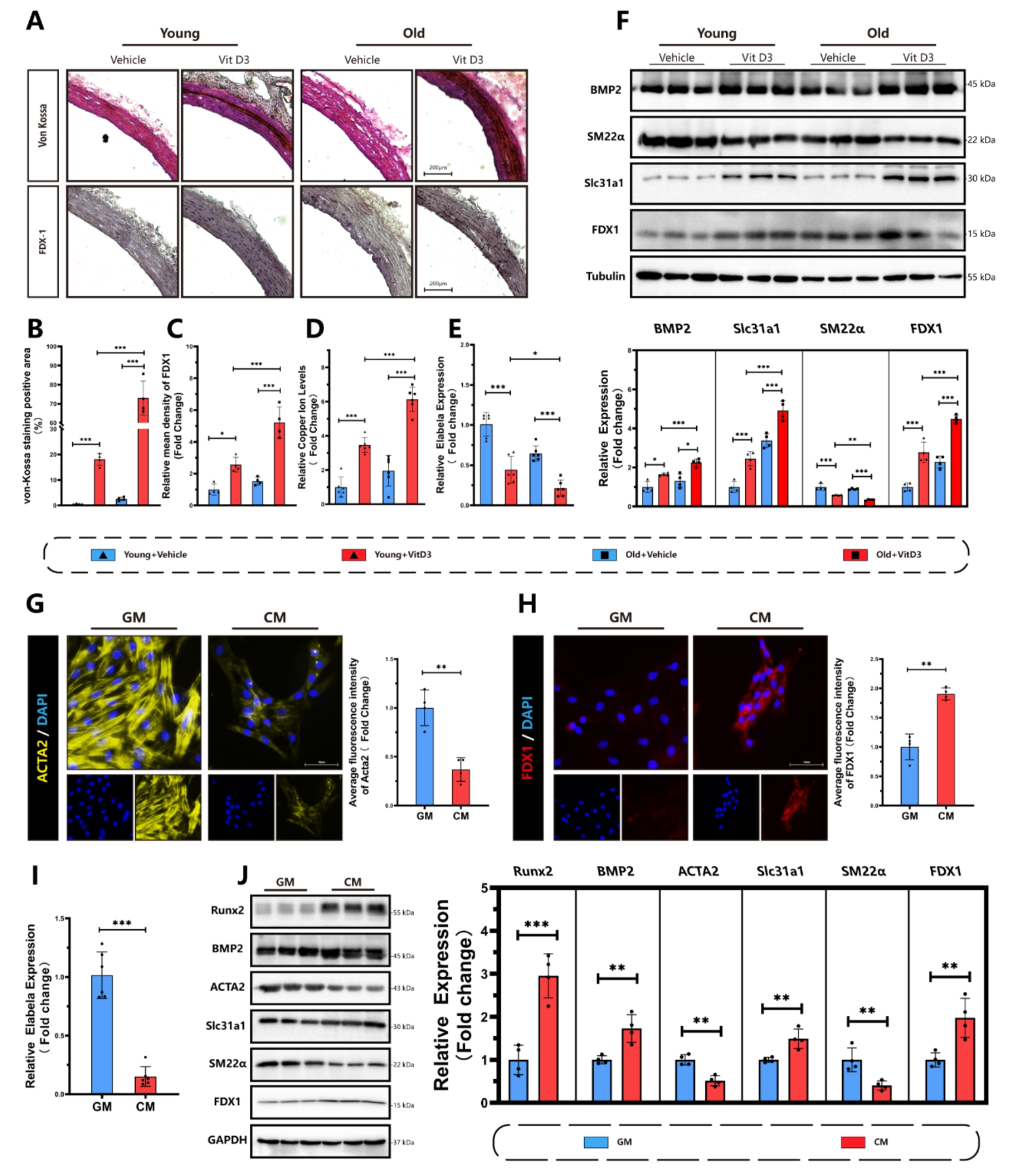
Mouse model of OVA‐induced ARThe mice were randomly divided into a control group (WT control group), an allergic rhinitis group (WT AR group), a Tet2−/− control group (Tet2−/− control group) and a Tet2−/− allergic rhinitis group (Tet2−/− AR group) each consisting of 10 mice. In the AR and Tet2−/− AR group, mice were sensitized by an intraperitoneal administration of 100 μg ovalbumin (OVA, grade V; Sigma-Aldrich, St. Louis, MO, USA) and 5 mg aluminum hydroxide in 300 μL normal saline on days 0, 7, and 14. The control group received an intraperitoneal injection of the same dose of saline. The mice were then intranasally challenged with 20 μL saline (10 μL per nostril) containing 200 μg OVA daily for 2 weeks starting on day 14. Similarly, in the control group, mice were intranasally challenged with the same amount of saline. In AR + Met group, in the local challenge stage, the metformin solution (250 mg/kg) was injected intraperitoneally to remove endotoxin 30 min before each challenge, and the other mice were the same as the AR group. Within 15 min after the last OVA challenge, the frequency of nasal rubbing and sneezing in each mouse was counted to quantitatively evaluate the symptoms of AR. Figure 1 provides a summary of the experimental procedure.
Fig. 1
Flow chart of mouse model and confirmation of successful establishment of AR model. A In AR group, 300 μL OVA was intraperitoneally injected at 0, 7 and 14 days after the start of the experiment to achieve basal sensitization. On days 21 to 35 of the experiment, local excitation was performed by intranasal infusion of 20 μL OVA nasal drops at 2 PM every day. On the 36th day of the experiment, behavioral experiments were prepared or sacrificed. Mice in control group were treated with normal saline instead of OVA intraperitoneal injection and OVA nasal drops. The model of Tet2−/− control group was the same as that of control group. Mice in Tet2−/−AR group were modeled in the same way as those in AR group. B Quantitative analysis of sneezing times in control group and AR group within 15 min. C Quantitative analysis of nasal scratching times within 15 min in control group and AR group. D Comparison of serum OVA-specific IgE levels between control group and AR group. E HE and PAS staining of mouse nasal mucosa (×400 times). Black arrows indicate eosinophils and red arrows indicate goblet cells. (***p < 0.001)
Measurement of OVA‐specific IgESerum levels of OVA-specific IgE were determined using a Mouse OVA-specific IgE ELISA Kit (Cayman, Ann Arbor, USA) according to the manufacturer's instructions.
Hematoxylin and Eosin (HE) stainingMice were anesthetized with 3% isoflurane inhalation and quickly decapitated. Coronal brain slices (300 µm) containing ACC were prepared by standard methods and tissues of bilateral ACC were dissected in ice-cold artificial cerebrospinal fluid (ACSF) under the anatomical microscope (Xu et al. 2008). Noses and ACC tissues were fixed in 4% paraformaldehyde for 48 h. Additionally, the noses were decalcified in decalcified solution for 2 weeks and then made into paraffin sections. After the paraffin sections were deparaffinized, hematoxylin was used to stain the nucleus and eosin was used to stain the cytoplasm. The morphology of the nasal mucosa and ACC tissues were then observed under a light microscope (×400). Five fields on each slice were randomly selected, in which the number of eosinophils was counted under the microscope and then averaged.
Periodic acid-Schiff (PAS) stainingThe nasal mucosal tissues were fixed in a 4% paraformaldehyde solution. Then, the paraffin-embedded tissue samples were cut into 4-μm-thick sections. The sections were stained with periodic acid-Schiff (PAS) stain for goblet cells. The numbers of these cells were counted in 4 randomly high-power fields (magnification 400×).
Nissl stainingMice were perfused transcardially with 200 mL of ice-cold phosphate-buffered saline, followed by 300 mL of phosphate-buffered 10% formalin. After perfusion, the brain was fixed for 48 h, dehydrated, and embedded in paraffin. Hippocampal tissues were collected prepared as 10-μm-thick sections and stained by a Nissl dye. The experiment was performed using a Nissl staining kit (Solarbio, China) according to the manufacturer’s instructions. Nissl-stained cells in the ACC areas were observed at ×400 magnifications.
Immunofluorescence stainingThe ACC tissues were fixed with 4% paraformaldehyde, embedded in slices, and baked. After the paraffin sections were completely dewaxed with xylene, 10% calf serum was added, and the section were placed at room temperature for 10 min. The sections were incubated with anti-NLRP3 (1:500, Proteintech), anti-Caspase-1 (1:1000, Proteintech), anti-ASC (1:1000, ABclonal), and anti-IBA-1 (1:1000, Abcam) at 4 °C overnight, followed by the goat anti-mouse or goat anti-rabbit secondary antibody for 1 h at room temperature. The sections were washed with water, blown dry, sealed with glycerin, and followed by observation under a fluorescence microscope (Olympus, Tokyo, Japan). For cellular immunofluorescence, BV2s were seeded into 6-well plates containing a coverslip per well at a density of 0.25–1 × 10^6, fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 10 min. After being rinsed with PBS, cells were blocked with 2% BSA and then incubated with anti-NLRP3 (1:200, Proteintech), anti-Caspase-1 (1:200, Proteintech), anti-ASC (1:200, ABclonal), and anti-IBA-1 (1:1000, Abcam) at 4 °C overnight, followed by the goat anti-mouse or goat anti-rabbit secondary antibody for 1 h at room temperature. Expression of various indicators in different groups on three high-power fields (HPF, × 400) were studied by a fluorescence microscope (Olympus, Tokyo, Japan).
Mouse PET experiment and software analysis procedureBefore PET imaging, mice were fasted for 12 h and then injected with approximately 200 ± 10 μCi of 18F-FDG (18-fluoro-6-deoxyglucose) through the tail vein. After 60 min of metabolism, mice were anesthetized with 2% isoflurane. TransPET Discoverist 180 (Suzhou Ray-can Technology Co., LTD) system was used for static scanning for 10 min to obtain images, and then CT images were scanned. PET images were reconstructed by (3D) OSEM with a 3D voxel size of 0.5 × 0.5 × 0.5 mm3, and CT images with a matrix of 256 × 256 × 256 were reconstructed by FDK algorithm. Region of interest (VoI) analysis was performed using Amide (Medical Imaging Data Review software) and Pmod (Pmod Technologies LLC, Zurich PET Center, Switzerland) software. The mean normalized uptake value (SUV) was calculated using the following formula: mean pixel value with decay corrected VoI (μCi/kg)/(injected dose [μCi]/ weight [kg]).
PET images were analyzed by software, ROIs were delineated, and SUV values of each ROI were calculated. The calculation method of SUV is shown in Eq. (1).
$$SUV = \frac }} }} \times \frac }} }} \times W_$$
(1)
CT is the activity in a unit volume of tissue. DInj is the injection dose. VT is the volume of tissue. WT is the mass of tissue. WS is the mass of mice. Here, the tissue density VT/WT is set to 1.
Behavioral experiment and specific stepsElevated cross maze experimentThe elevated cross maze mainly consists of a pair of open and closed arms that are perpendicular to each other and cross each other, connected in the middle. Data were collected by a surveillance camera located above the center of the experimental area, and processed by a computer animal behavior analysis software connected to the camera. The equipment size of mouse elevated cross maze: arm length 70 cm, arm width 5 cm, closed arm height 15 cm, arm height 50 cm off the ground.
Before the experiment, make sure that the whole device of the elevated cross maze is clean. In the experiment, the mice were gently removed from the cage with their backs to the experimenter as far as possible, and placed gently in the central area of the organ body, with the mice facing the open arm. Then the experimenter left quickly and quietly, clicked the start button of the software, and recorded the activity of the mice within 5 min. ETHOVISION animal movement trajectory tracking system was used to record and analyze the behavior of each mouse in the elevated cross maze experiment. Anxiety behavior was evaluated by the time the mice entered the open arm, and the longer the time they stayed in the closed arm, the more anxious the mice were. If any mouse remained stationary in the closed arm or fell from the open arm, the experimental data were excluded.
Open field testThe open field experimental apparatus consists of mouse open field experimental chamber and data acquisition and processing system. The specification of mouse experimental chamber is (length × width × height): 50 cm × 50 cm × 40 cm.
Before the experiment, it is necessary to ensure that the whole device of open field experiment is clean. In the experiment, the mice were gently removed from the cage as far away from the experimenter as possible and placed gently in the central area of the open field. Then the experimenter left quickly and quietly, clicked the start button of the software, and recorded the activity of the mice within 5 min. ETHOVISION animal motion trajectory tracking system was used to record and analyze the behavior of each mouse in the open field experiment. The total movement distance of mice was used to reflect the movement situation and reflect the spontaneous activity ability of mice. The anxiety and depression of mice were reflected by the residence time of central zone and the total times of entering central zone.
Tail suspension testRemove the experimental animal gently from the cage, cut out 17 cm long electrical tape (1–1.5 cm wide), and mark 2 cm at one end. The marked part of the tape was connected to the tail of the mouse, leaving a distance of about 3 mm from the tail, and the remaining 15 cm of tape was used for hanging the mouse. Attach the tape to the hanging rod, so that the abdomen of the mouse faces the camera (to capture the movement of the limbs), and leave immediately; the activity of animals in the air was recorded for a total of 6 min. The first immobile time in the first 2 min and the accumulated immobile time in the next 4 min were recorded. The earlier the immobility time in the first 2 min, the more likely the animals were to induce depression. The longer the immobility time within the next 4 min, the more depressed the animals were.
Forced swimming testBefore the experiment, it is necessary to ensure that the whole device of the forced swimming experiment is clean. Then, tap water is added to the cylindrical water tank with a bottom radius of 6 cm and a height of 30 cm, the temperature is controlled at about 25 ℃ (the upper and lower levels are not more than 1 ℃), and the liquid level is adjusted to about 18 cm. In the experiment, mice were gently removed from their cages and quickly placed in water. At the same time, the activity of the mice was recorded for a total of 6 min. The cumulative immobility time of the animals after 4 min was counted. The longer the immobility time, the more serious the depression was. At the end of the experiment, the mice were put into the clean cage prepared in advance, and the animals were assisted to recover their body temperature. The next batch of experiments were conducted after changing water and cleaning.
Sucrose preference testIn brief, the mice were habituated to 1% sucrose solution for 1 h. Two bottles with 150 mL 1% sucrose solution or water were offered to mice. The bottles were weighted prior to or following the test. The mice were fed freely before the experiment, and tap water and sugar water consumption were measured by measuring the weight of the bottle. After calculating the consumption of water and sucrose solution, the formula was applied for determining the sucrose preference: sugar water preference value = sugar water consumption (g)/[sugar water consumption (g) + water consumption (g)] × 100%.
Western blot analysisThe mouse tissues and BV2 cell lines were homogenized and lysed in RIPA buffer with added protease and phosphatase inhibitors to extract the total protein. The protein concentration was measured using a BCA Protein Assay Kit (Beyotime Biotechnology, China). Protein samples totaling 40 μg were added to each lane of the gel, separated on a 10% SDS-PAGE gel, and then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore) for 1.5 h at 200 mA. The PVDF membranes were sealed with 5% skim milk for 1 h at room temperature. The membranes were washed three times for 5 min each and then incubated with anti-TET2 antibody (1:1000, Proteintech), anti-NLRP3 (1:500, Proteintech), anti-Caspase-1 (1:1000, Proteintech), anti-ASC (1:1000, ABclonal), and anti-IBA-1 (1:1000, Abcam) antibody overnight at 4 °C. The membranes were washed three times with TBST for 10 min each and then incubated with the goat anti-mouse or goat anti-rabbit secondary antibody for 1 h at room temperature. The membranes were visualized by chemiluminescence using an Image Lab™ quantitative assay system (Bio-Rad, California, USA).
Dot-blot assayLevels of 5-hmC in the ACC were detected using dot-blot assays as described previously (Ko et al. 2010). Briefly, genomic DNA was isolated from brains of WT and AR mice. The DNA samples were denatured and dilution samples were spotted on nitrocellulose membranes. The blotted membrane was vacuum-baked at 70 °C for 1 h, blocked, and incubated with anti-5-hmC antibody (1:10000, CST) overnight at 4 °C. After being incubated secondary antibody, the membrane was visualized by Image Lab™ quantitative assay system (Bio-Rad, California, USA).
Enzyme-linked immunosorbent assay (ELISA)The ACC tissues, blood and culture media were collected after drug treatments. The ACC tissues on both sides of a mouse were taken, and 200 μL of lysate was added. After homogenization, the lysate was split on ice for 30 min and centrifuged at 12,000 rpm for 15 min. The protein concentration of the supernatant was measured by a BCA kit (Absin, Shanghai, China), and ensured that the total protein of each sample was 50 µg. Similarly, supernatants of blood and culture media were retained. The levels of inflammatory mediators were measured using ELISA kits of IL-1β, IL-18 (BD Biosciences, San Jose, CA, USA) and OVA-specific IgE (Bioswamp, Wuhan, China) according to the manufacturer's instructions, respectively. Briefly, 50 μL ELISA diluent and 50 μL sample were added to each well, and incubated at room temperature for 2 h. After aspirating and washing 5 times, 100 μL of the working detector was added and incubated at room temperature for 1 h. Then, 100 μL of TMB one-step substrate reagent was added and incubated at room temperature for 30 min. After adding 50 μL stop solution, read at 450 nm within 30 min. The experiments were repeated for 3 times.
siRNA and plasmid transfectionBV2 microglia cells in culture flasks were seeded at a density of 5 × 105 cells in each 6-well plate, shaken evenly, and gene knockdown could be carried out when the cell density reached about 30% under a light microscope. siRNAs against TET2 (5'-CTGCTTCTGTTCTCAATAA-3') were obtained from GenePharma Co. (Shanghai, China). siRNAs or plasmids were mixed with Lipofectamine 2000 (Invitrogen, Carlsbad, USA) in reduced serum medium (Opti-MEM; Gibco, USA) according to the manufacturer’s instructions. Transient transfections were carried out as described previously (Qin et al. 2022). The medium was replaced for the cells in the plate during the resting period. After 15 min, 200 μL mixture was added to each well, and 6-well plates were labeled, gently mixed, and placed in the incubator. After 24 h, the concentration and state of the cells were observed, and the fluorescence expression after transfection was observed under a fluorescence microscope if there was a fluorescent label. Then 1 mL of complete medium was added to each well to supply the cells with energy, and the cells were placed in an incubator for 12–24 h.
Statistical analysisAll results are represented as mean ± standard error of mean. The data and graphs were analyzed by GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). The results were analyzed by 1-way analysis of variance followed by post hoc Tukey's tests for multiple comparisons. A P value of < 0.05 was considered significant.
Comments (0)