
Remember me
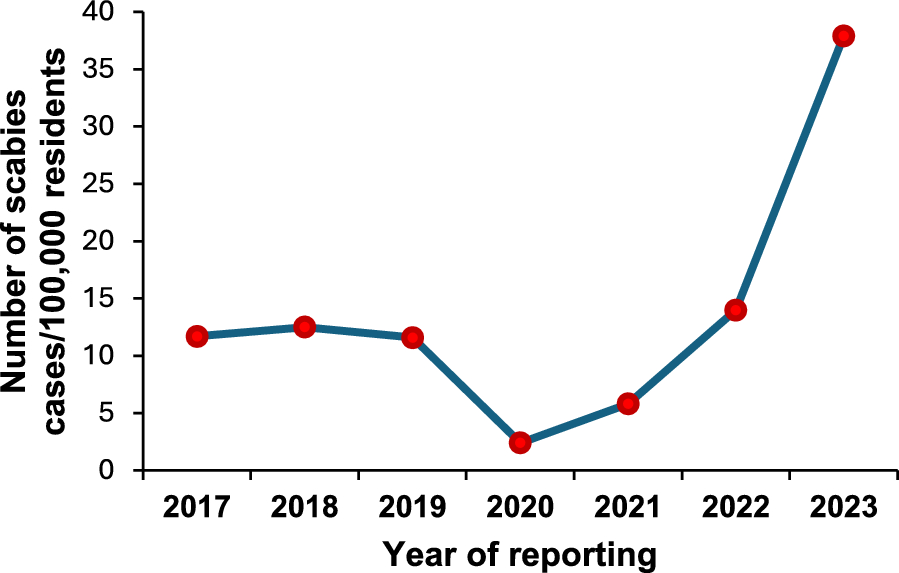
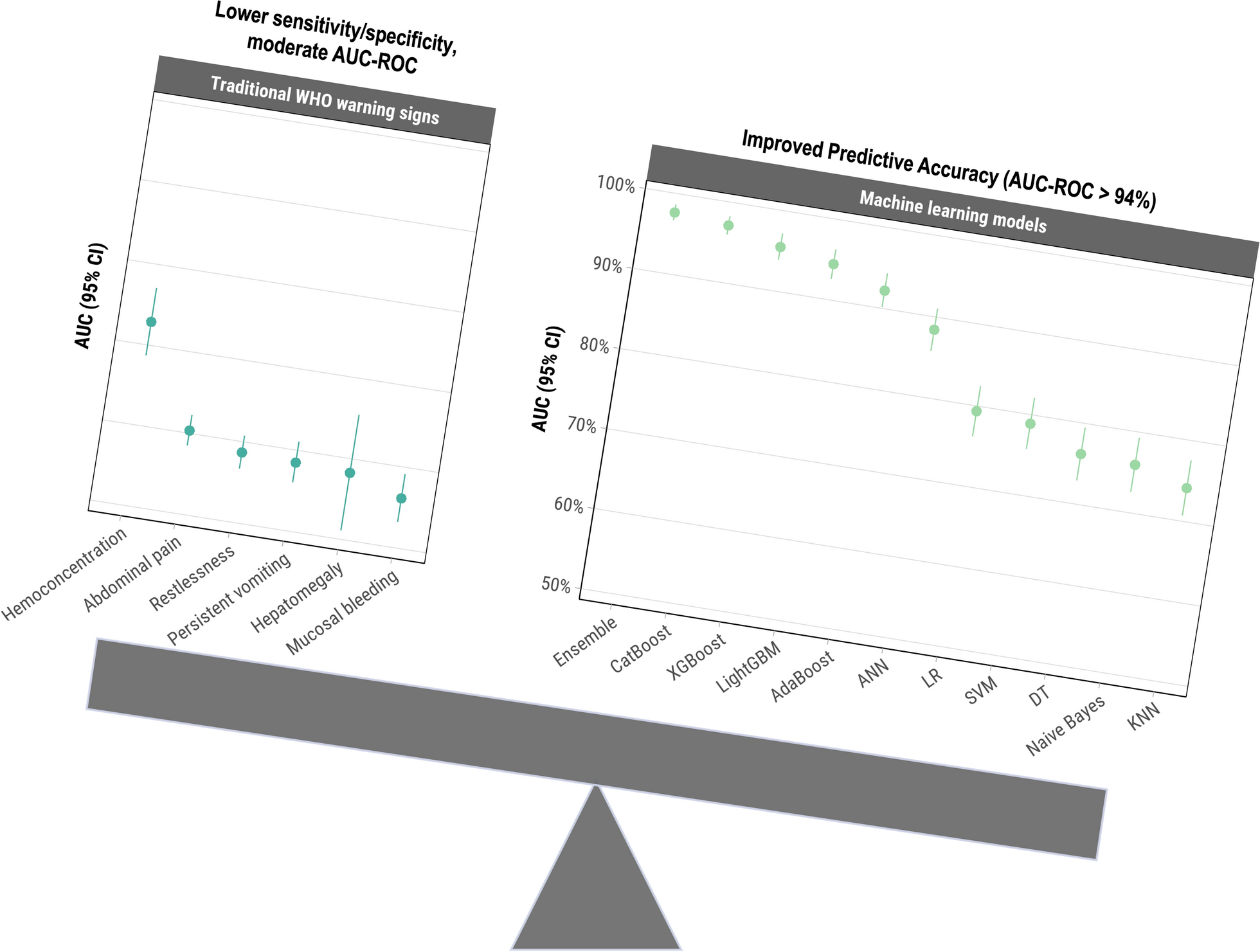
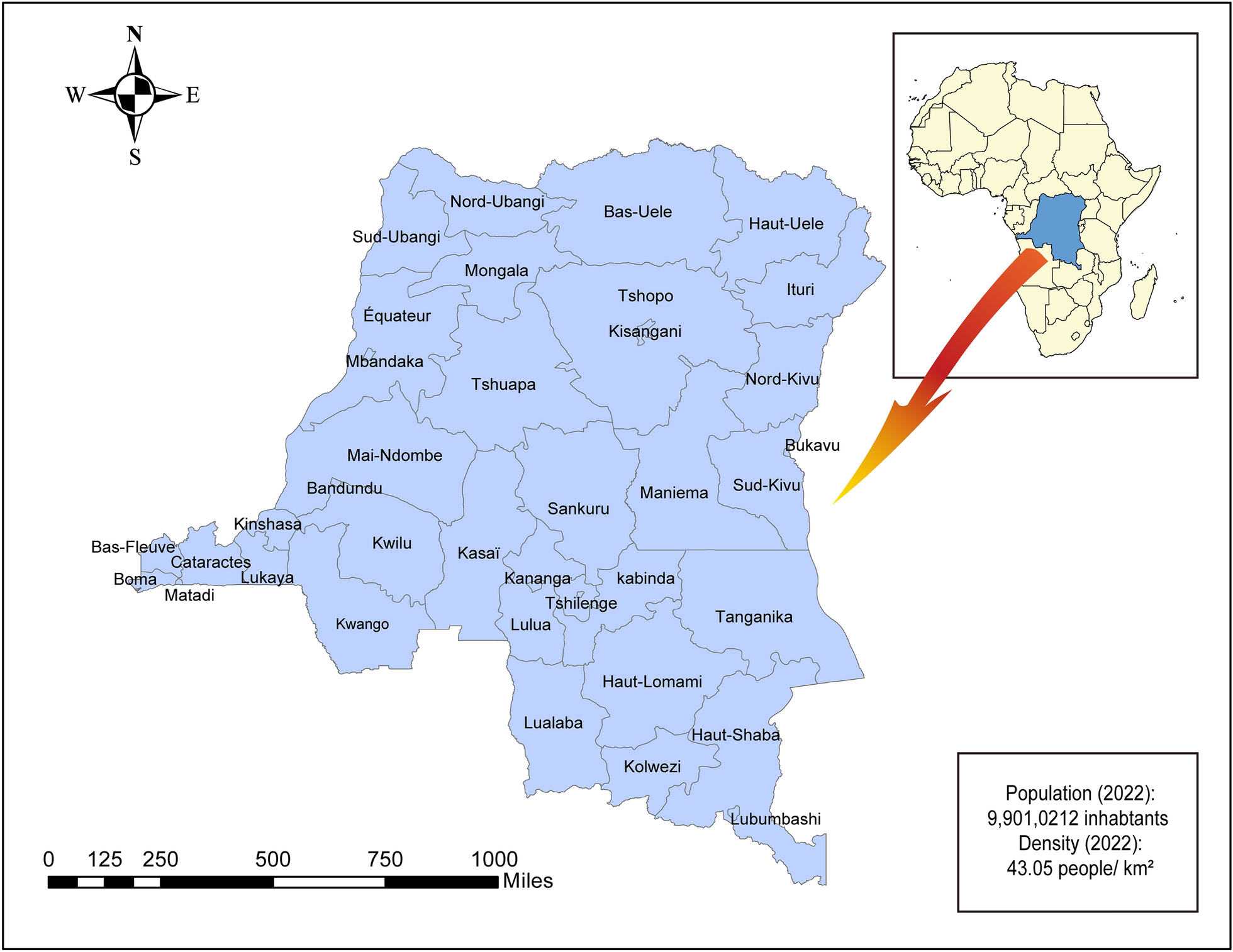
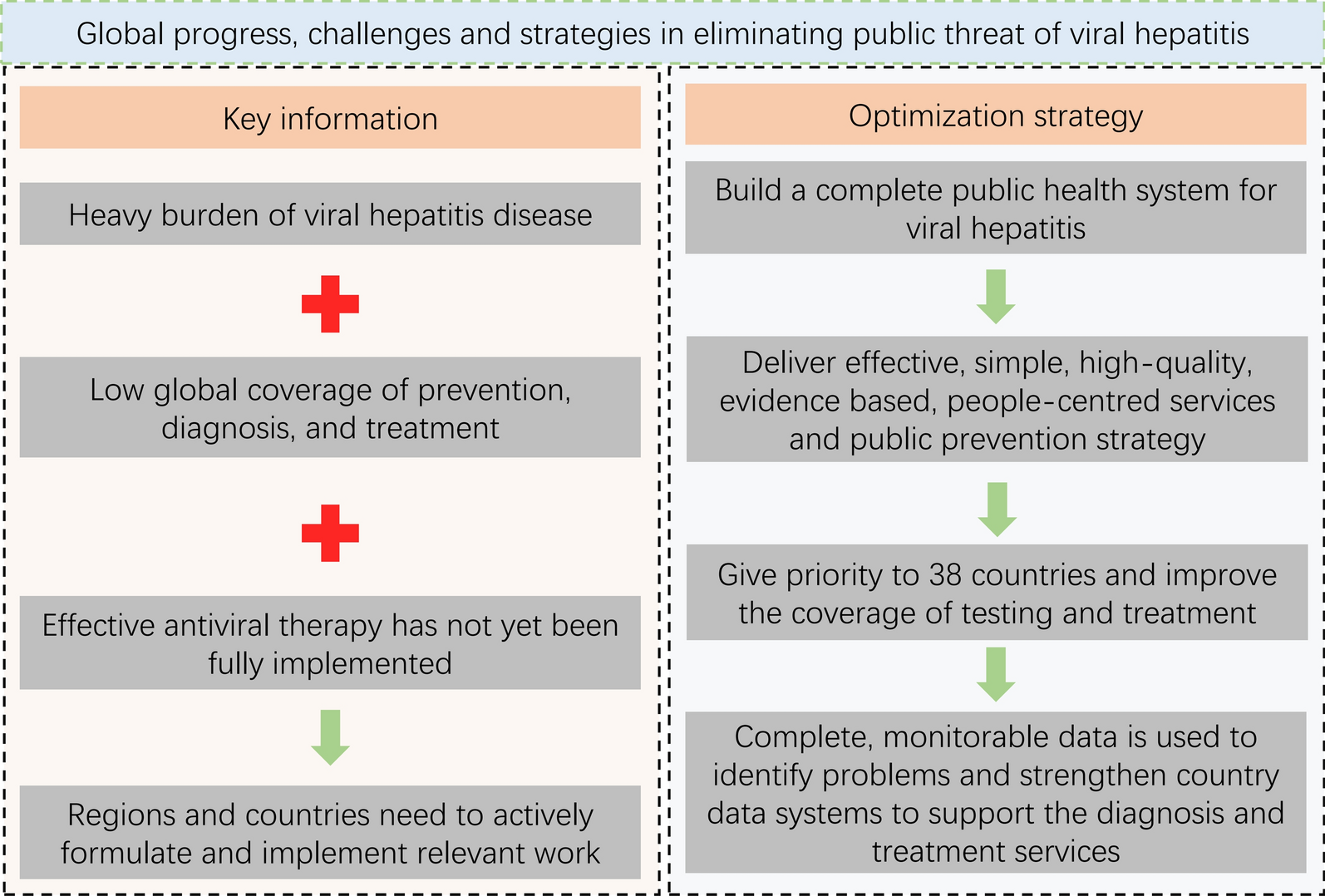




The sample populations in this study were divided into three groups. Group 1 was assigned for evaluation of urinary OV-RDT performance: well-defined faecal-positive and -negative individuals for O. viverrini (n = 493) from endemic areas in northeast Thailand (samples were obtained from the Department of Parasitology Biobank, Khon Kaen University, Thailand). The sample population in group 2 was selectively recruited for cross-reactivity assessment of urinary OV-RDT with other helminthiases identified by faecal examination (n = 96). The sample populations in group 3 were individuals participating in an ongoing prospective field trial and a treatment follow-up study on opisthorchiasis. These participants were residents from subdistricts of Khon Kaen, Maha Sarakham and Roi Et provinces of northeast Thailand with a known high prevalence of O. viverrini infection (n = 1629) (Table 1). The study was conducted from March 2020 to December 2022.
Table 1 Sample populations employed for diagnostic accuracy and cross-reactivity studies (group 1–2) and field testing of urinary OV-RDT (group 3)Clinical sample collection and processingAll participants provided demographic data and clinical specimens. Both faecal and urine samples were collected on the same day. For faecal samples, 2 g of fresh stool was preserved in 10% formalin and processed by FECT as a reference test. Urine samples were kept on ice for transportation to the laboratory at Khon Kaen University and were centrifuged at 500×g for 15 min at 4 °C to separate supernatants and stored at − 20 °C for analysis by urinary OV-RDT and ELISA.
Formalin-ethyl acetate concentration technique (FECT)The quantitative FECT was used for faecal examination in this study because it has advantages over other methods in using fresh or formalin-preserved faecal sample, it facilitates the discrimination of O. viverrini and minute intestinal flukes (MIF; Phaneropsolus/Prosthodendrium) and it is sensitive for light infections as well as for Strongyloides infections [12, 25]. For quantitative diagnosis, FECT yielded faecal egg counts comparable to Kato-Katz method and correlated with worm burden, and also antigen concentration in urine [22, 24,25,26]. The procedures for quantitative FECT were used as described previously [21]. Briefly, 2 g of fresh or preserved stool samples were homogenized, filtered, and centrifuged, and ethyl acetate was added for extraction of fat from the stool. The faecal suspension was centrifuged at 500 × g for 5 min, and the supernatant was discarded and the resulted sediment fixed with 1 ml of 10% formalin. The final faecal suspension was examined with three drops of 40 µl per sample using a compound microscope. The mean number of eggs was multiplied by the number of drops in the suspension, and divided by the mass of stool in grams to calculate the number of eggs per gram of faeces (EPG) [14].
Antigen preparationIn this study, the crude somatic antigen from adult O. viverrini was used. The antigen extraction procedure previously published was followed [27]. In brief, grossly undamaged worms obtained from the biliary system of infected hamsters were washed with sterile phosphate buffer saline (PBS) pH 7.2 and homogenized in glass tissue grinder at 4 °C in a small volume of PBS in the presence of 1 × Protease inhibitor Cocktail (Calbiochem, CA, USA). The worm homogenates were ultrasonicated and kept overnight at 4 °C before being centrifuged at 5000×g for 30 min. The supernatant was used as somatic antigen and kept in small aliquots at − 20 °C. The protein contents were measured by the Bradford’s method [28].
Mouse immunizationFive-week-old male BALB/c mice were immunized a total of 3 times with 25 μg (200 μl) of crude O. viverrini at a concentration of 0.5 mg/ml in an equal volume of Freund’s complete (first immunization) or incomplete (second and third immunizations) adjuvants administered subcutaneously at three-week intervals. The final immunization was undertaken with 25 μg of crude O. viverrini in normal saline solution (NSS). Serum samples were collected prior to the immunizations for the analysis of antibody titers. Mice were housed in the specified pathogen-free animal facility at Northeast Laboratory Animal Center, Khon Kaen University.
Hybridoma productionThree days after the last immunization, mice were sacrificed to remove the spleens for single cell preparation. A single cell suspension was prepared by pressing the spleen with syringe plug that was placed over a metal mesh in serum-free RPMI 1640 medium on a sterile petri dish, and carefully disaggregated using a cell strainer to form a single-cell suspension. The spleen cell suspension was washed three times with cold serum-free RPMI 1640 medium and centrifuged at 400 × g at 4 °C for 5 min followed by 3 washes using a serum-free medium (SFM) (Thermo Fisher Scientific, Waltham, MA, USA). The cells were mixed at a ratio of 1:5 viable parental myeloma cells to each viable splenocyte, and carefully fused using 1 ml of polyethylene glycol (PEG) (Sigma-Aldrich, St. Louis, MO, USA) for 1 min. The fused cells were resuspended in SFM and incubated in a water bath at 37 °C for 15 min, serum containing medium (SCM; 10% of fetal bovine serum in SFM), placed in a T-75 cm2 tissue culture flask (Sigma-Aldrich, St. Louis, MO, USA) containing 20 ml of 10% SCM (total culture volume is 30 ml), and incubated at 37 °C and 5% CO2 overnight. The fused cell suspension was removed from the flask, centrifuged, and transferred to a bottle containing 90 ml of ClonaCell™-HY Medium D (STEMCELL Technologies, Waterbeach, Cambridge, UK) and mixed thoroughly. The Medium D-containing fused cells were carefully transferred to the Petri dishes (1 × 10 cm). The dishes containing fused cells were incubated at 37 °C and 5% CO2. After 14 days, colonies detected on each Petri dish were transferred into an individual well of a 96-well tissue culture plate (Sigma-Aldrich, St. Louis, MO, USA) containing 200 μl of selection media (5% of fetal bovine serum in SFM with 1 × hypoxanthine, aminopterin, and thymidine (HAT; Sigma-Aldrich, St. Louis, MO, USA), and the plates were incubated at 37 °C and 5% CO2 for 3–4 days prior to preliminary screening by indirect ELISA.
Screening of hybridoma cells by indirect ELISAScreening of monoclonal antibody clones was performed by indirect ELISA. The flat-bottomed microtiter plates (Maxisorp, NUNC, Denmark) were coated overnight at 4 °C with 100 μl/well of 5 μg/ml crude O. viverrini in carbonate buffer pH 9.6. The plates were washed three times with normal saline containing 0.05% Tween 20 (NSST) and were blocked with 200 μl/well 5% skim milk in carbonate buffer pH 9.6 at 37 °C for 1 h. The plates were then washed as above, and 100 μl/well of hybridoma culture medium was added after which the plate was incubated at 37 °C for 1 h. Following washing, bound antibodies were detected by the addition of 100 μl/well of HRP-conjugated rabbit anti mouse IgG (Invitrogen, CA, USA), diluted at 1:2000 in 2% skim milk in PBS containing 0.05% Tween 20 at 37 °C for 1 h. After washing, 100 μl/well of 0.04% OPD in citrate phosphate buffer pH 5.0 was added and incubated at RT for 20 min. The reaction was stopped with 100 μl/well of 4 mol/L H2SO4 and the plates were read at 492 nm. Pre-immunization mouse sera and the sera from mice that received the full course of immunization (100 μl, diluted 1:1000 in PBST) were used as negative and positive controls, respectively.
Production and purification of monoclonal antibodiesThe antibody isotype of the final hybridoma cell line (OV-3) was determined by a Pierce Rapid ELISA Mouse mAb isotyping Kit (Invitrogen, CA, USA). The isotyping results showed that the clone was the mouse immunoglobulin subclass 2b (IgG2b) with kappa as the light chain. The monoclonal antibody was produced by in vitro culture of the hybridoma cell line. It was adapted to hybridoma serum-free media (HSFM) (Invitrogen, CA, USA) and cultured in a 1000 ml WHEATON® CELLine™ flask (Taylor Scientific, St. Louis, MO 63144, USA) using HSFM. The highly concentrated monoclonal antibody was harvested, and fresh media was added for media replacement. The viability of hybridoma cells was determined by trypan blue dye exclusion assay. Culture supernatants were pooled and were purified using a HiTrap® IgG Purification HP (GE Healthcare Bio-Sciences, Uppsala, Sweden) attached to an AKTATM start chromatography system (GE Healthcare Bio-Sciences, Uppsala, Sweden) following the manufacturer’s instructions. Briefly, the cell supernatant was precipitated with saturated (NH4)2SO4 to a final concentration of 0.8 mol/L. The ammonium sulfate-containing supernatant was filtered through a 0.45 µm filter immediately before applying it to the column. Before applying the sample to the column, the column was washed with 5 ml of distilled water to remove ethanol, then the column was equilibrated with 5 ml of binding buffer [20 mmol/L sodium phosphate and 0.8 mol/L (NH4)2SO4 with pH 7.5] at a flow rate of 1 ml/min. The sample was applied to the column at a flow rate of 1 ml/min. Eluted and collected the bound proteins (IgG) with 10 ml elution buffer. The unbound sample was washed out using 15 ml of binding buffer with a flow rate of 1 ml/min until the absorbance reached a steady baseline. After elution, the column was regenerated and washed with 7 ml of wash buffer (20 mmol/L sodium phosphate, pH 7.5 with 30% isopropanol) and re-equilibrated with 5 ml of binding buffer, prior to the subsequent purification. Finally, the IgG isolated was kept at -80 ℃ for urine ELISA and urinary OV-RDT.
Monoclonal antibody-based urinary antigen ELISAThe protocol for O. viverrini urinary antigen measurement by ELISA was slightly modified from previous descriptions [21, 22]. The main modifications were the use of monoclonal antibody cell line (OV-3) and shortened incubation times to 45 min for urine samples, captured rabbit IgG antibodies and biotinylated anti-rabbit IgG conjugates, while the rest of the procedures remained the same.
Known cases of O. viverrini infection-negative and -positive urine samples (n = 20) determined by faecal FECT were used to construct a receiver operation curve (ROC). The cut-off points (OD value was 0.31) for diagnosis by urine ELISA was calculated using MedCalc software version 9.6.3 (MedCalc, Ostend, Belgium). The standard curves for the calculation of antigen concentrations in urine samples were constructed based on spiked urine with crude antigen of O. viverrini by ELISA. The relationships between the urinary concentrations (X) and OD values (Y) from ELISA were estimated by the best-fit linear regression equation of Log Y = 0.767X − 0.854. From the cut-off of OD = 0.31, the calculated cut-off value of OV antigen in urine became 32.9 ng/ml.
Opisthorchis viverrini rapid diagnostic test for opisthorchiasis (urinary OV-RDT)The structure of OV-RDT was based on immunochromatographic lateral flow methodology, which is standard with in vitro diagnostic medical devices (IVDs). The test cassette consisted of a series of membranes, test (T) and control (C) lines. The key component is the specific monoclonal antibody to O. viverrini antigen (clone OV-3), and the reaction is signalled by a nanogold particle anti-mouse IgG conjugate. OV-RDTs were produced in collaboration with the National Center for Genetic Engineering and Biotechnology, and K Bioscience Ltd, Thailand. The mAb and anti-mouse IgG (Lampire Biological Laboratories, Pipersville, PA, USA) were dispensed onto nitrocellulose membranes (Sartorius Stedim Biotech SA, Goettingen, Germany) to serve as the T and C lines, respectively. The optimal conditions were as follows: 1 mg/ml of goat anti-mouse IgG was absorbed at the control line, 2 mg/ml of mAb was absorbed at the test line, and 10 μg/ml of mAb conjugated with colloidal gold was sprayed onto a glass microfiber filter GF33 (conjugate pad) (Whatman Schleicher & Schuell, Dassel, Germany) (Fig. 1).
Fig. 1
Schematic diagram of urinary OV-RDT for the diagnosis of opisthorchiasis (A). OV-RDT interpretation and grading score (B). mAb monoclonal antibody, OV-RDT Opisthorchis viverrine-rapid diagnosis test. The color intensity at T was expressed as + 4 for the highest intensity and + 1 for the lowest intensity
The test was performed by applying three drops of urine sample (120 μl) and one drop of buffer (40 μl) in the receiving bay then allowing the reaction to occur over 10 min to obtain test results. For interpreting the results, if there were two reactive bands of both T and C lines, the test was positive for opisthorchiasis. If there was one band at the control line, the test was negative for opisthorchiasis. The control line should always appear, indicating test validity. The interpretation of the urinary OV-RDT result was based on the intensity of T-band. The grading scores from 2 independent assessors were compared to finalize the results. The inconsistencies of faint/indecisive band were considered negative. The standard OV-RDT colour chart for the grading score based on the band intensity from 1–4 was constructed by using O. viverrini crude antigen diluted in clean urine beginning from 5000 ng/ml (+ 4) and diluted to 28.5 ng/ml (+ 1) (Fig. 1).
Limit of detection (LOD) for urinary antigen ELISA and OV-RDTCrude somatic antigens were spiked in negative urine specimens by starting from 10 µg/ml to 5000 µg/ml for LOD analysis by ELISA and urinary OV-RDT.
Statistical analysisData were analysed by using SPSS version 22 (International Business Machines, USA). The McNemar chi-square test was used to determine the significance of differences in proportions for prevalence by diagnostic testing. The Kruskal–Wallis tests were used to assess the correlation between antigen concentration and the grading score of urinary OV-RDT. The performance of OV-RDT for sensitivity, specificity, and predictive values was calculated by using faecal FECT as a reference standard. Analysis of diagnostic agreements between tests was measured by Cohen’s kappa coefficient (κ-value) and was interpreted using kappa value guidelines: κ < 0 indicated no agreement, 0 to 0.2 indicated poor agreement, 0.21 to 0.4 indicated fair agreement, 0.41 to 0.6 indicated moderate agreement, 0.61 to 0.8 indicated good agreement, and 0.81 to 1.0 indicated excellent agreement [29]. Statistical significance was reached when P values were > 0.05.
Comments (0)