
記住我
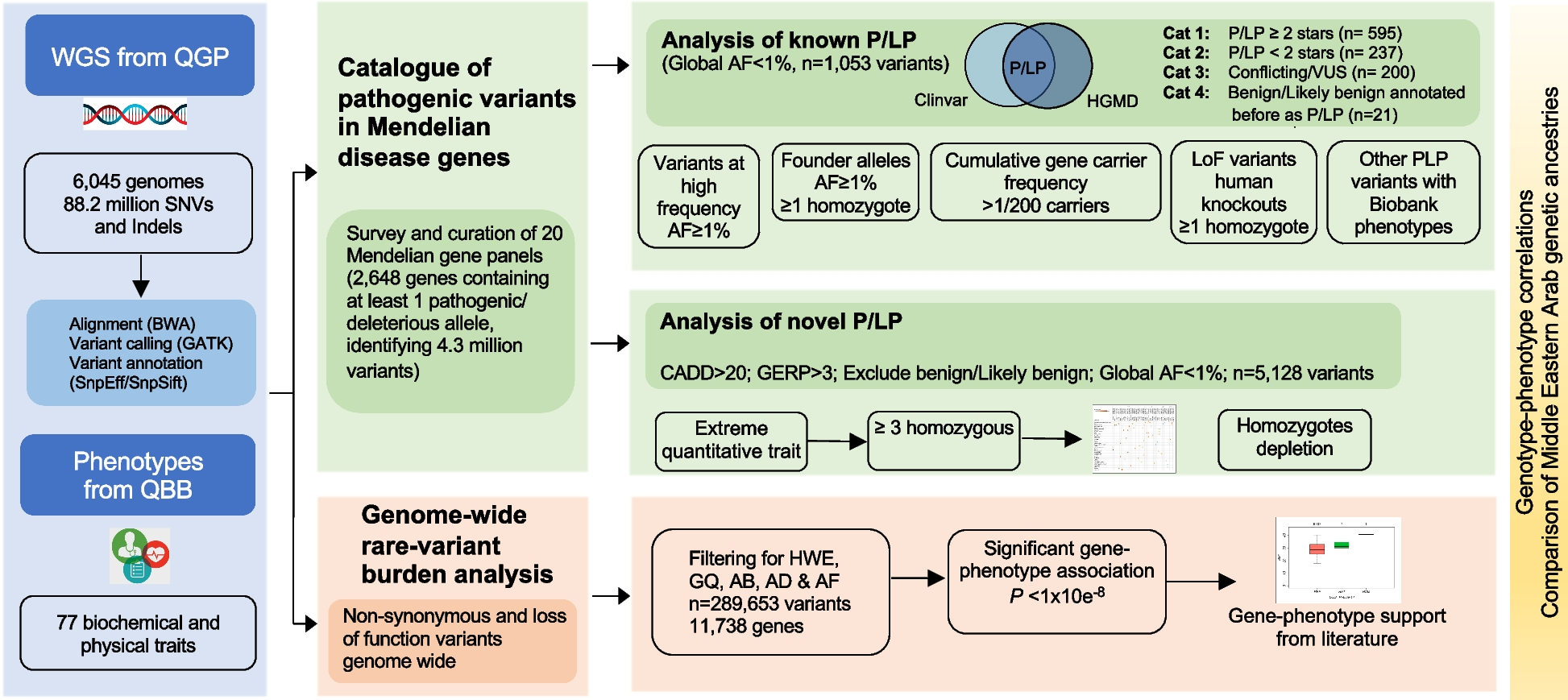





Similar to previous results at the NOD2 locus [16], across all 11 Sazonovs et al. [10] risk genes, and 4 protective genes, rare variants inferred to be causally related to Crohn’s disease in European ancestry (EA) individuals are one-fifth as prevalent in African Americans (AA). All comparisons were significant by Fisher’s exact test. Figure 1 shows this graphically for all 25 variants, with data summarized in Table 1. Reduced minor allele frequency (MAF) was observed consistently for two common risk variants in EA with MAF > 0.05 at HGFAC and SLC30A8, for six rare risk variants with 0.01 < MAF < 0.05 at CCR7, DOK2, SDF2L1, and NOD2 (all four are well-established coding mutations), and a total of 11 very rare or ultra-rare risk variants with MAF < 0.01, one each at RELA, PTAFH, PDLIM5, IL10RA, and seven at NOD2. Concordantly, six rare variants in IL23R, TYK2, TAGAP, and CARD9 that are protective in EA were even more rare in AA. In all cases, the difference was replicated in two AA cohorts (our IBD-GC case-control WGS cohort and gnomAD AA) and three EA datasets (UK Biobank, Sazonovs et al., and gnomAD Non-Finnish Europeans).
Fig. 1
Minor allele frequencies of rare variants across European and African cohorts. Bars show MAF of the 25 rare variants reported to be causal for IBD by Sazonovs et al. [10], grouped by risk or protective status, with colors representing the cohorts shown in the legend at bottom. The SNP ID coordinates are shown for GRch38 with corresponding rsIDs from dbSNP; rs IDs for the ultra-rare NOD2 SNPs are from left to right the same order as top to bottom in Table 1
Table 1 Allele frequencies and effects of rare coding variants affecting CD susceptibilityAs shown in Table 2, we also identified 4 variants in these loci with MAF > 0.01 in AA which have low evolutionary probability, namely EP-scores in the range of those of known pathogenic variants, but are nearly absent in Europeans: one in SDF2L1, two in PTAFR, and one in IL10RA. The latter variant may be protective since the minor allele frequency is 4.5% in both the AA-WGS controls and gnomAD African ancestry sample, but just 4.1% in the AA-WGS cases. The other three are more likely risk variants since they are slightly elevated in the AA-WGS cases (but also in the gnomAD African ancestry sample). A further rare variant in PTGER4, the gene with the highest common variant effect specifically in African Americans [22], may also be a risk allele in African ancestry individuals alone since it is also absent from Europeans, though it too is also slightly higher frequency in gnomAD.
Table 2 Variants in Sazonovs et al. loci in African Americans, with EP-scores within pathogenic range (EP < 0.0022)Variance explained by established rare causal variants for Crohn’s disease in African AmericansDespite the five-fold reduction in MAFs between European and African IBD and control cohorts, evaluation of allelic effect sizes is necessary to establish whether they also explain a considerably lower proportion of the risk of Crohn’s disease in African Americans. Power to establish whether a variant is a risk factor or not is low as there were only 1774 AA cases and accordingly only R161H in SDF2L1 was even nominally significant (p = 0.005) in AA, yet all 8 risk alleles with European MAF > 0.01 are concordant in their observed direction of effect in AA and EA (contrast the heights of the dark and lighter-blue shades for AA, or dark and lighter reds for the two EA cohorts in Fig. 1). Furthermore, the computed odds ratios are slightly higher for half of the variants as documented in Table 1. Regarding the protective alleles, 3 of 4 with measurable effects are also nominally protective in African Americans, the exception being P1104A in TYK2.
Table 1 shows the variance explained in our IBD-GC AA WGS cohort and the Sazonovs et al. cohorts [10], computed as 2pq(lnOR)2 where p is the MAF and q = 1-p. Excluding the 11 very- or ultra-rare variants for which odds ratio computation is unreliable, averaged across the other 5 risk alleles not including NOD2, the rare coding variants discovered in Europeans are predicted by this method to have similar population attributable risk in African Americans as in Europeans, cumulatively 1.39%. However, more than half of this is due to R161H in SDF2L1, which may have an inflated estimate due to sampling variance, while the variants in HGFAC and SLC93A8 also make appreciable contributions. The three established major risk factors at NOD2 by contrast explain just 0.75% of the variance in AA, compared with up to 12% in EA. Regarding the protective variants, the lower allele frequencies result in much less protection in AA, an estimated 1% versus 7.6% in EA, although most of the latter is due to the relatively high frequency of R381Q in IL23R in EA.
An alternative mode of measuring the burden of rare variants is to calculate the difference between the observed number of cases and that expected if the odds ratio of each variant were 1. Cumulatively, the 19 risk variants were observed in 405 of the 1744 cases, and 318 of the 1644 controls in the AA WGS dataset, for a combined odds ratio of 1.26. For comparison, the mean odds ratio in Europeans, excluding NOD2, was 1.41. If we assume for the sake of direct comparison a prevalence of 1% in both African and European ancestry populations, this implies 41 excess cases per 100,000. This estimate includes the very rare variants, but since their contribution is small, restricting the analysis to the 8 rare variants above also yields a similar reduction in burden in African Americans as compared to European ancestry. Not accounting for co-occurrence of multiple variants in some individuals and summing the individual excess burden results in 59 cases per 100,000 in AA and 271 in EA from Table 1. Correspondingly, we estimate 15 versus 111 fewer cases in AA and EA respectively, due to the 6 documented protective rare variants. Cumulatively, then, the 25 rare Sazonovs et al. variants are expected to contribute to the occurrence of 44 excess cases of Crohn’s disease per 100,000 African Americans, four-fold fewer than the 160 excess cases (1 in 625) in European ancestry individuals. This mode of analysis thus agrees with the interpretation based on percent variance explained.
Most of the known European-discovered rare variant burden for Crohn’s Disease in African Americans is due to admixtureTo determine whether the lower allele frequencies in African ancestry individuals may reflect lower nucleotide diversity at these IBD risk loci in general, we computed coding region nucleotide diversity (π) on African and European-derived chromosomes (see Methods; Additional file 1: Fig. S1). There was no consistent pattern, with four loci showing elevated diversity and three reduced diversity on African-derived haplotypes and three with similar measures. Similar results were observed for the four loci with rare protective variants. Since genome-wide diversity is known to be ~30% greater in African than European populations, most of these genes have higher than expected diversity in Europeans relative to Africans [14], possibly indicating reduced selection outside Africa.
The Sazonovs et al. variants allele frequencies were evaluated in West Africans from the 1000 Genomes database; however, because their AFs were nearly 0, we did not include these estimates in Fig. 1. All 8 of the rare risk alleles are at a lower frequency in West Africans in the 1000 Genomes database than in our AA WGS cohort study. At NOD2, the R702W MAF is close to 1% in both West Africans and the gnomAD AA cohorts, whereas G908R and 1007fs are absent. Four of the other 5 variants are also absent or nearly so in West Africans in the 1000 Genomes database, the exception being rs16844401 in HGFAC, which has a MAF of 2.3%, compared with 6.5% in non-Finnish Europeans. Just one of the protective alleles, rs41267765 in TAGAP, has a MAF greater than 1% in Africans, and it is the only variant in the dataset predicted [36] to pre-date human origins.
These results suggest that the presence of most of the Sazonovs et al. variants in African Americans may be predominantly due to admixture. To confirm this, we used Gnomix [31] to paint each of the chromosomes on which the genes are found, allowing inference of the genetic background of origin for each variant in each individual. Strikingly, as seen in Fig. 2A, at CCR7, DOK2, SLC39A8, and NOD2, just 17/300 risk alleles (less than 5%) appear to be on African haplotypes. In addition, all 14 of the 15 very or ultra-rare variant instances at four genes (RELA, PTAFR, PDLIM5, and IL10RA) are European-derived. At HGFAC and SDF2L1, the two loci with common risk variants, the proportions of African and European-derived risk variants are close to 50%, which is still well below the genome-wide average of non-pathogenic alleles. The protective variants at IL23R, TYK2, and TAGAP are also mostly European-derived, to varying degrees. As a control for these inferences, we show in Additional file 1: Fig. S2 that the major alleles follow the expected distribution for African Americans of ~80% African ancestry, in both cases and controls, reflecting the known contribution of admixture to African ancestry genetic proportions.
Fig. 2
Admixture proportion of rare IBD variants. A Proportion of copies from African (blue) and European (brown) haplotypes containing Sazonovs et al. [10] inferred causal variants for carriers in the African American WGS cohort. B Proportion of copies from African and European haplotypes containing Sun et al. rare variants [24] associated with different disease groups for carriers in the African American WGS cohort. The numbers above each bar represent the total number of copies in both A and B
A corollary of these results is that there is a highly significant correlation between the proportion of the genome, at the 13 chromosomes, derived from European ancestry, and risk of CD. Figure 3A shows that for the 19 causal risk variants, fraction of European ancestry increases with number of causal rare variants (p = 1.2 × 10–9), slightly more in the cases than controls. The ideograms in Fig. 3 panels C and D show admixture segments by chromosome for a control and case individual respectively, each with 4 causal variants (gold arrowheads) where the European ancestry (pink intervals) is clearly greater in the case. Gnomix [31] overestimates the number of breakpoints due to error in phasing, but this should not affect the ancestry inference at most loci.
Fig. 3
Relationships between admixture proportion and genetic risk. A Genome-wide fraction of European ancestry per individual against total number of Sazonovs et al. variants carried per individual. B PRS derived from African weights (blue) and European weights (red) vs. fraction of African ancestry. C Chromosome painting for an African American control carrying four inferred causal Sazonovs et al. variants. D Chromosome painting for an African American IBD case carrying four inferred causal Sazonovs et al. variants. The blue and pink colors in the chromosome paintings represent African and European, respectively
We also asked whether African ancestry proportion correlates with polygenic risk score for the common variant risk for IBD. Using a PRS generated using liability-scale allelic effect weights computed in the UK Biobank for 215 established common risk variants, as reported in [22], a weak but highly significant negative correlation (r = − 0.16, p < 10–16) between African ancestry proportion and PRS was observed for the AA cases in the IBD-GC WGS dataset. This appears to be because there is a slight excess of risk variants that are more common in Europeans, so admixture tends to increase the proportion of risk variants. Substituting effect weights estimated for African Americans markedly increased the correlation (r = -0.42) as seen in Fig. 3B. This is likely due to 55% of the risk variants having larger effect sizes observed in AA, increasing the contribution of admixed variants to the PRS. It also suggests that the slightly elevated prevalence of cases in the top percentiles of risk in AA with this AA-weighted PRS [22] is actually due to increased admixture, noting a small but significant correlation between proportion of European ancestry and IBD prevalence in the IBD-GC cohort. Presumably, there are also common variants yet to be discovered in poorly sampled African ancestry populations that would produce opposing effects on overall risk but are not captured by the existing PRS.
Similar biases are observed for other common complex diseasesSun et al. [24] reported 975 coding-wide significant variants for a wide range of diseases in the initial release of the UK Biobank WES data supplemented with FinnGen WES. We evaluated the relative burden in the gnomAD African and NFE cohort subsets using the same approach as for the IBD risk loci, but without differentiating between cases and controls. Across all loci, there was a highly significant difference in frequency of pathogenic rare variants (MAF < 0.05), with an average MAF of 0.003 in AA and 0.017% in NFE (t-test, p = 2 × 10–16). Figure 2B shows that just 8 of 105 variants detected in the IBD-GC WGS cohort were observed on African-derived haplotypes, consistent with the majority being present due to admixture. These are partitioned by disease category in Fig. 2B, and reduction in rare variant allele frequency was significant for the loci associated with diseases of the circulatory system, sense organs, and endocrine/metabolic system, and trending for congenital abnormalities, digestive, genitourinary, hematopoietic, musculoskeletal, mental, and respiratory disorders. We emphasize that in each case, the evaluated variants were discovered by association studies carried out in a very large European ancestry cohort study and that comparative discovery data for AA populations is not yet available.
留言 (0)