Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the major cause of dementia. Progressive memory loss and cognitive decline are the hallmarks of AD [1]. It was assessed that AD affects 40 million individuals globally, and this number is anticipated to double every 20 years until at least 2050 [1]. In addition to ageing (ages ≥ 60 years), other risk factors of AD, include familial background, cardiovascular changes, traumatic brain injury, malnutrition, various infectious agents, psychiatric factors and metabolic disorders, such as diabetes [2], [3]. Neuropathological features of AD include extracellular deposits of amyloid-β (Aβ) peptide, and neurofibrillary tangles made up of intraneuronal fibrillar clusters of hyperphosphorylated Tau proteins. The persistent inflammation driven by Aβ peptide deposits, and overexpression of inflammatory mediators around the Aβ plaques as well as neurofibrillary tangles are related to neurodegeneration in AD [4].
Amyloid precursor protein (APP), a transmembrane molecule, is normally involved in the control of neuronal development and differentiation. In AD, β- and γ-secretase degrade overexpressed APP to produce Aβ peptides, which cause neurotoxicity after aggregation [5]. Some AD forms are due to mutation in genes encoding APP, tau, presenilins (PS1 and PS2), β- and γ-secretase [6], [7]. In order to mimic some human AD features, various mouse models expressing mutants of human APP, tau and other related genes have been developed [6], [7]. These models can differ in terms of pathological process, amyloid deposit burden, formation of neurofibrillary tangles, affected brain regions, neuronal and synaptic loss, and onset time [6], [7].
Triple transgenic AD (3xTg-AD) mice, for example, carry three mutants, including human APP Swedish, tau and presenilin mutants [8], [9]. 3xTg-AD mice, Aβ peptide deposits appearing from six months of age and tau-related pathologic reactions beginning in the hippocampus and spreading to the neocortex by 10–12 months of age (49). At 3 months of age, microglial activation together with enhancement of inflammatory markers occur in this model [10]. Because this model reflects the main pathogenic characteristics of the human AD. It has been extensively used in the research of AD pathogenesis and in the evaluation of potential treatment approaches [6].
To accelerate plaque development, the researchers engineered a transgenic mouse model that co-expressed 5 mutant genes (5xTg-AD). 5xFAD mice carrier five, including 3 mutations (Swedish, Florida, and London mutations) in the APP gene, and 2 familial AD (FAD)-related mutations in the PSEN1 gene (encodes PS1) that promote the formation of Aβ peptide deposits [7]. In addition to powerful gliosis, Aβ peptide plaques in the brain develop more quickly at two months of age and expand in an age-dependent process and eventually reaching saturation by six to nine months of age (51). It should be mentioned that, unlike 5xTg-AD mice, no AD patient was found to carry all five familial mutations [11].
APP/PS1 mice harbor the Swedish mutation and another mutation in the FAD-linked PSEN1 gene. At six months of age, APP/PS1 exhibit Aβ peptide deposits which reach to large amounts in the cortex and hippocampus by nine months [7]. In this model, reactive astrocytes surround the Aβ peptide deposits [7]. As a model of Aβ-induce AD, APP/PS1 mice exhibit Aβ peptide deposits inducing AD-like pathology, whereas in 3xTg-AD mice both Aβ deposit- and tau-related pathological processes contribute to the disease development.
It should be noted that the appropriate folding of amino acids that make up a protein is a prerequisite for its optimal activity [12]. In order to maintain optimal functional activities, cells have the ability to effectively control protein synthesis and assembly and establish protein homeostasis (proteostasis). Deficiencies in proteostasis as well as protein misfolding and self-aggregation cause cell dysfunction and apoptosis [12]. Insoluble aggregates formed by the misfolded proteins can exert cytotoxicity effects [12]. The progressive deposition of misfolded proteins, without their adequate removal can cause amyloid disease, the most frequent of which is AD [12].
Ageing is a crucial risk factor for neurodegenerative diseases. Aging is related to massive and progressive misfolding and aggregation of proteins in different tissues. Misfolded aggregates are more likely to develop and accumulate in older cells and tissues [13]. One of the main features of many age-related protein misfolding diseases (especially different neurodegenerative diseases like AD) is the formation of misfolded protein aggregates [13]. In addition to amyloid deposition, sustained inflammation secondary to misfolded protein can contribute to AD pathogenesis [14]. Age-related declines in proteasome activity impair cellular to eliminate damaged proteins which promotes disease development [13], [15]. Inhibition of autophagy also exacerbates protein aggregation. In a mouse AD model, heterozygous deletion of an autophagy-regulating gene (namely beclin 1) hampered autophagy aggravating AD pathogenesis [13].
If misfolded proteins are not sufficiently eliminated, they can enter into peripheral lymphoid tissues, where they are presented to naïve T cells via antigen-presenting cells. After that, naïve T cells undergo differentiation into antigen-specific Th1, Th2, Th17, CD8+ CTLs, and Treg cells. Th1 and Th17 cells, in particular, are infiltrated into the CNS and cause neuroinflammation directly. CD8+ CTLs identify antigens that are presented by MHC class I on the nervous cells and trigger neurotoxicity [16]. Self-aggregated/misfolded proteins can disrupt immunological tolerance by inducing autoreactive effector T cells and decreasing Treg cell-mediated anti-inflammatory neuroprotective activities [14]. On the other side, innate immunity as well as adaptive immunity have the ability to trigger neuroinflammation and hasten the formation of aggregated/misfolded proteins that cause neuronal death [14].
Microglia, astrocytes, infiltrated immune cells, cytokines and chemokines can contribute to AD-related neuroinflammation. In the early stage of AD, immune system activation and phagocytosis might aid in the clearance of Aβ peptides and stop the development of amyloid plaques. Chronic inflammation, on the other hand, may become harmful because, if the amyloid burden cannot be cleared, it can contribute to the development of AD [17]. Systemic inflammation may also affect the pathogenesis and progression of AD. Systemic inflammation disrupts the blood–brain barrier (BBB), allowing peripheral leukocytes to enter the brain [18]. AD patients express greater circulatory amounts of cytokines, such as IL-1β, IL-6, TNF-α, and TGF-β [18]. In the early stages of AD, the Aβ peptides can drain from the brain to peripheral lymph nodes, where they activate specific T cells that eventually enter the CNS [19], [20], [21]. As the illness progresses, meningeal lymphatic activity declines, blocking the Aβ peptide drain, which increases peptide accumulation in the meninges [22].
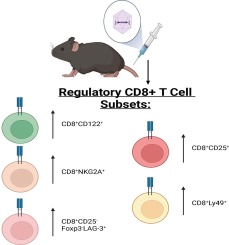
Immune cells, in particular, the effector T lymphocytes of the immune system were divided into several subsets, including Th1, Th2, Th9, Th17, Th22, and regulatory T (Treg) cells playing various roles during neuroinflammation [23]. Although Treg cells exert anti-inflammatory and immunosuppressive effects, their precise role is complex, and there are controversies regarding their numbers and functions in AD patients as well as in related animal models. This review aimed to provide a comprehensive figure regarding the role of Treg cells in AD while highlighting potential therapeutic strategies.









留言 (0)