Antibodies and tetramers
Cetuximab (CTX) and pembrolizumab were obtained from Merck (Germany, Darmstadt). Trastuzumab (TRS) and all genetically modified antibodies were produced at Genmab via transient expression in ExpiHEK293 FreeStyle cells as described before [23] and purified by Protein A affinity chromatography. If required, protein aggregates were removed via Size Exclusion Chromatography to yield protein product with a > 95% monomeric content as analyzed on HPLC-SEC. All antibodies were stored in phosphate-buffered saline (PBS). For the AECs used in vivo experiments mutations were introduced (P329G, L234A, and L235A) to disrupt possible interactions with Fc-receptors [24].
The following antibodies were used for flow cytometry: cetuximab, trastuzumab, goat anti-human IgG-A488 (Jackson immunoResearch, UK, Cambridgeshire, #109-546-098) or -PE (Jackson immunoResearch, #109-116-098), goat anti-mouse-FITC (Jackson immunoResearch, 115-096-062), mouse anti-HLA-A2 (produced in-house from clone BB7.2 [25]), mouse anti-human EGFR (Santa Cruz Biotechnology, #sc-120), and mouse anti-human Her2 (R&D, UK Abingdon, #MAB9896). Peptide MHC multimers complexes (tetramers) were generated in-house as described before [26] and labeled with PE-conjugated streptavidin.
Cell lines and cell culture
All adherent cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Massachusetts, Waltham), 1% Pen/Strep (Gibco), 10% Fetal Calf Serum (FCS, Biowest, France, Nuaillé). The U266 cells were cultured in IMDM (Lonza, Switzerland, Basel), 10% FCS (Gibco, USA, Massachusetts, Waltham), 3 mM L-glutamine (Lonza, Switzerland, Basel), 1% Pen/Strep. HeLa cells were transduced with the aid of a pEF1α lentiviral vector encoding the cDNA of HLA-A2 or GFP as a Mock. In the HeLa-HLA-A2 (HeLa-A2) cell line EGFR and Her2 knockout lines were generated as described before [22]. The U266 cell line and one of the HeLa-A2 knockout Her2 clones were transduced with MP71 retroviral vectors encoding an intracellular domain truncated Her2 or EGFR receptor (tHer2 or tEGFR). Cell cultures were enriched for transduced cells by FACS sorting using an Aria III cell sorter (BD Bioscience, Germany, Heidelberg).
The T-cells used were specific for EBV-BRLF1 (YVLDHLIVV presented in HLA-A*02:01), or CMV-pp65 as a non-specific control (NLVPMVATV presented in HLA-A*02:01) and were either previously established T-cell clones or CD8+ T-cells derived from peripheral blood mononuclear cells (PBMCs) transduced with the virus-specific TCRs. T-cells were cultured in T-cell medium (TCM); IMDM (Lonza, Switzerland, Basel), 5% FBS (Gibco, USA, Massachusetts, Waltham), 5% human serum (Sanquin, the Netherlands, Amsterdam), 3 mM L-glutamine (Lonza, Switzerland, Basel), 1% Pen/Strep, and 200 IU/ml IL-2, and stimulated every 10-16 days with phytohaemagglutinin (PHA, ThermoFisher, Germany, Dreieich) and irradiated feeder cells. Before the T-cells were used in the assays cells were washed 3 times with IMDM supplemented with 0.5% human albumin (Albuman, Sanquin) to remove expansion-related cytokines. Mycoplasma tests were performed regularly by PCR and were negative throughout the duration of the experiments.
TCR identification and TCR gene transfer to CD8+ T-cells
TCRs of selected T-cell clones were sequenced [27]. The TCR chains were codon optimized, synthesized, and cloned in a MP71-TCR flex retroviral vector by Baseclear (the Netherlands, Leiden). The MP71 flex retroviral vector contains codon-optimized and cysteine-modified TCRαβ (mTCR) constant domains to optimize TCR expression and increase preferential pairing [28]. Phoenix-AMPHO cells were transiently transfected with the generated constructs and virus supernatant was harvested and stored at −80 °C. From PBMCs, CD8+ T-cells were isolated by Magnetic-activated cell sorting (MACS) with CD8+ isolation beads (Miltenyi Biotec) and stimulated with PHA with irradiated autologous feeder cells at a 1:3 ratio. On day 2 after stimulation, 24 well suspension plates were coated with retronectine (Takara, France, Paris) and blocked with 2% human serum albumin (HSA). Next, the retroviral supernatant was added, and cells were spun down for 20 min at 4 °C, 2000 × g. The retroviral supernatant was removed, and the activated T-cells were transferred to the wells. After overnight incubation, the T-cells were transferred to a new culture plate. On day 7 after stimulation the TCR positive cells were MACS enriched using an anti-mouse TCR-Cβ-APC (mTCR, BD Pharmingen, Germany, Heidelberg) antibody in combination with anti-APC beads (Miltenyi Biotec). On day 8–10 after stimulation the T-cells were analyzed on a FACS for CD8, and mTCR expression, and transduced T-cells were used in experiments or re-stimulated to expand further.
Flow cytometry
For flow cytometry experiments, 100,000 cells were plated in a 96-wells U-bottom plate, washed with PBS containing 0.5% bovine serum albumin (BSA) and 0.02% Natrium Azide (PBA), and incubated with primary antibody for 30 min on ice. Hereafter, the cells were washed 2x with PBA, followed with 20 min incubation with the secondary antibody. The cells were washed once and analyzed on a LSRII (BD, USA, New Jersey, Franklin Lakes). The amount of cell surface receptors was quantified for the different cell lines, using the QIFI kit (Agilent Dako, USA, Santa Clara) according to the manufacturer’s instructions.
T-cell activation assays
For the T-cell activation assays with adherent cell lines, 5000 target cells/well were cultured overnight in a 384-well flat-bottom tissue culture plate to allow them to adhere. Antibody dilutions were prepared in IMDM supplemented with 0.5% HSA and titrated concentration of the different antibodies were added to the adherent cells and incubated for 1 hr at 37 °C. The wells were washed 3x and subsequently 4000 T-cells/well were added to the adherent target cells in IMDM supplemented with 0.5% HSA and 100 IU/ml IL-2. Before the T-cells were added, they were washed 3x to remove expansion-related cytokines. After overnight coculture, IFN-γ production by the T-cells was measured in the supernatant by ELISA as a measure for T-cell activation (Diaclone, France, Besançon).
For the target cell lines in suspension, the tumor cells were exposed to the AECs diluted in IMDM supplemented with 0.5% HSA for 1 hr at 37 °C. Next, cells were washed 3x with IMDM supplemented with 0.5% HSA, and 40,000 cells/well were transferred to a 384-well flat-bottom tissue culture plate to which 4000 T-cells/well were added in IMDM supplemented with 0.5% HSA and 100 IU/ml IL-2.
T-cell cytotoxicity measurements
After harvesting supernatants from the cocultures of AEC incubated adherent cell lines and T-cells TCM was added and the cocultures were incubated for an additional 48 hrs. Subsequently, T-cells were removed by gentle washing (3x), and DMEM culture medium supplemented with 10% AlamarBlue HS cell viability reagent (ThermoFisher, USA, Massachusetts, Waltham) was added. Viability was measured in relative fluorescence units (RFU) according to the manufacturer’s protocol. The percentage target cell killing was calculated using:
$$ }=100-\frac}\,}-}\,}\,})}}\,}\,}-}\,}\,})} 100$$
In which RFU sample is the measured value of our samples, the average RFU background is the average value that comes from four wells in which only T-cells were cultured, and RFU no addition is the measured value of a coculture of target and T-cells without antibody or peptide treatment.
To measure T-cell-mediated cytotoxicity of the different U266 cells, the U266-tEGFR and U266-tHer2 were incubated for 1 hr with 100 nM of AEC at 37 °C. After removal of the AEC by 3 centrifugation steps with IMDM with 0.5% HSA, the U266 cells were cultured at 37 °C for 18 hr. Then the U266 cells were incubated 1 hr at 37 °C with 100 μCi 51chromium (PerkinElmer, USA, Massachusetts, Waltham), washed 3 times, and incubated at different E:T ratios for 7 h in 96-well U-bottom culture plates. The spontaneous release was measured by addition of only IMDM with 0.5% HSA and the maximum release by addition of 1% Triton X-100 (Sigma–Aldrich, USA, Missouri, Saint Louis). The supernatants were harvested and transferred to LumaPlates (PerkinElmer) and 51Cr release was measured on a 2450 Microbeta2 plate counter (PerkinElmer) in counts per minute (CPM). The percentage killing was calculated with a similar formula as for the Alamarblue assay.
In vivo experiments
Animal procedures were performed according to AVD116002017891 appendix 2 which was approved by the Central Committee of Animal Experiments (CCD, The Hague, The Netherlands) according to the European legislation (EU 2010/63/EU) and Animal Experimental Committee of Leiden University.
Female NOD-scid-IL2Rgammanul (NSG) 7–14 weeks old mice were injected intravenously (i.v.) with 2 × 106 U266 cells transduced with the extracellular domain of Her2 (tHer2) and Luciferase. After 14 days, the mice were injected i.v. with 5 × 106 T-cells transduced with the virus-specific TCR, followed by an i.v. injection on day 15 and 18 of the TRS-H (100 μg) diluted in PBS or an intraperitoneal (i.p.) injection of 100 μg on day 14 and 18 of CTX-H as a single treatment or in combination with 100 μg pembrolizumab. Tumor outgrowth was measured at regular intervals after subcutaneous (s.c.) injection of 150 μL 7.5 mM d-luciferine (Cayman Chemical) using a CCD camera (IVIS Spectrum, PerkinElmer). On the indicated days, blood was collected through a tail-vein puncture, and serum was collected and frozen. Group sizes were determined bases on variation observed in previous experiments and mice were randomized over groups based on the tumor outgrowth as measured on day 14. Treatment was not blinded.
Statistical analysis
Graphpad Prism software (V.9.0.1) was used to perform the statistical analysis. In the figure legend the used test is indicated, and the significance levels are indicated as ns as not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.


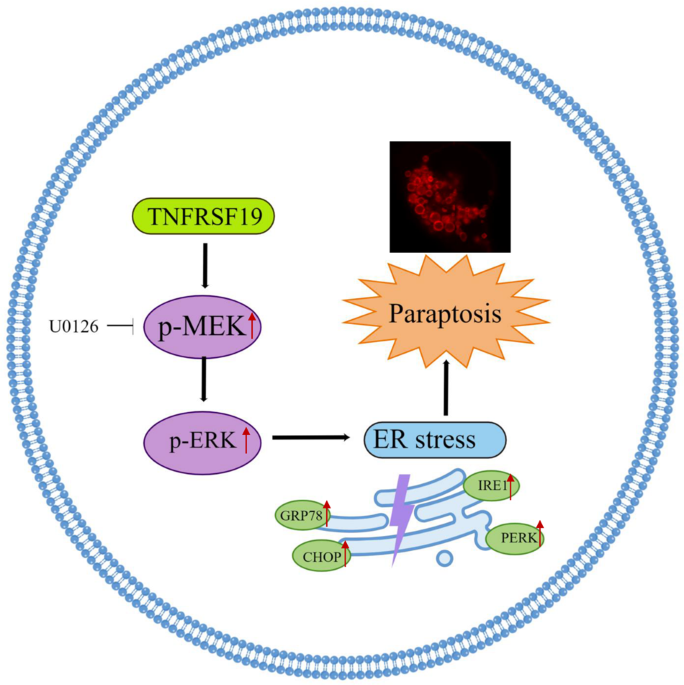
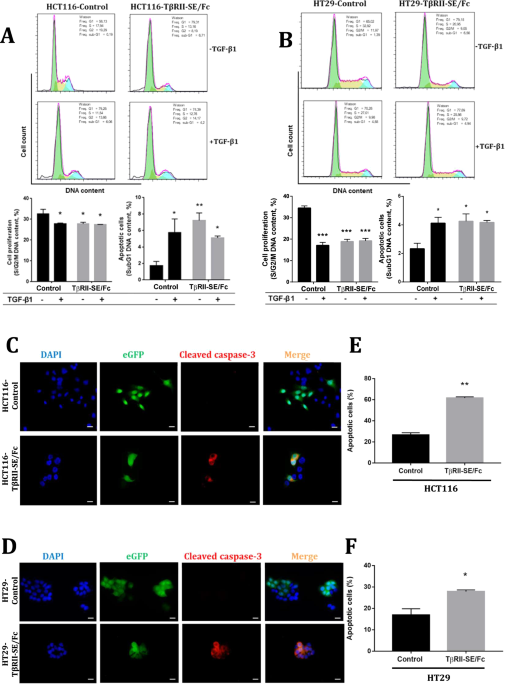
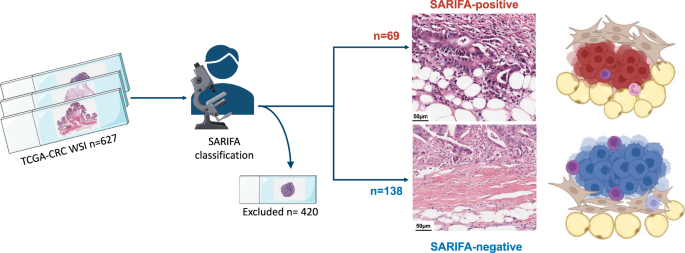
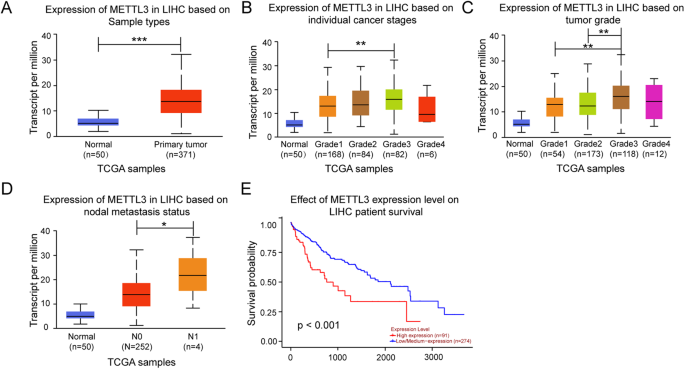
留言 (0)