
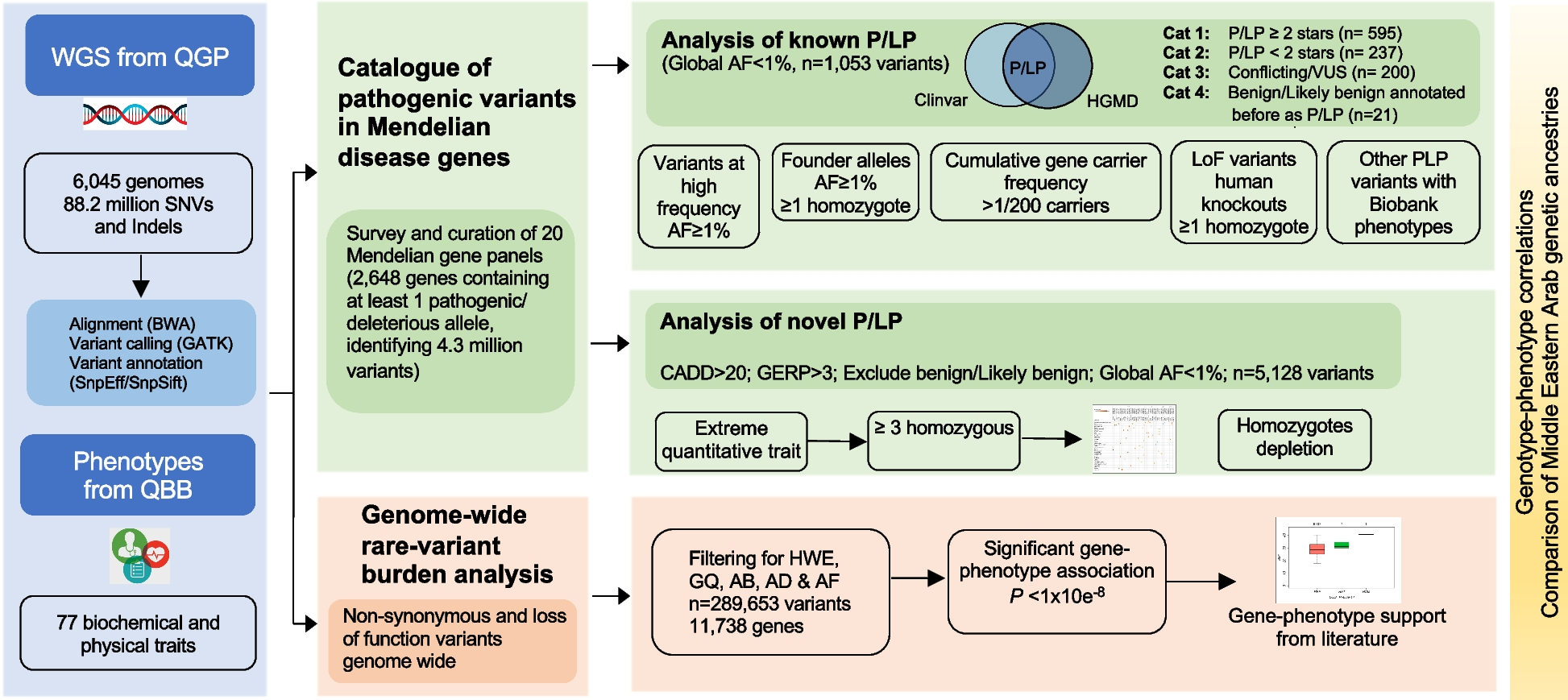
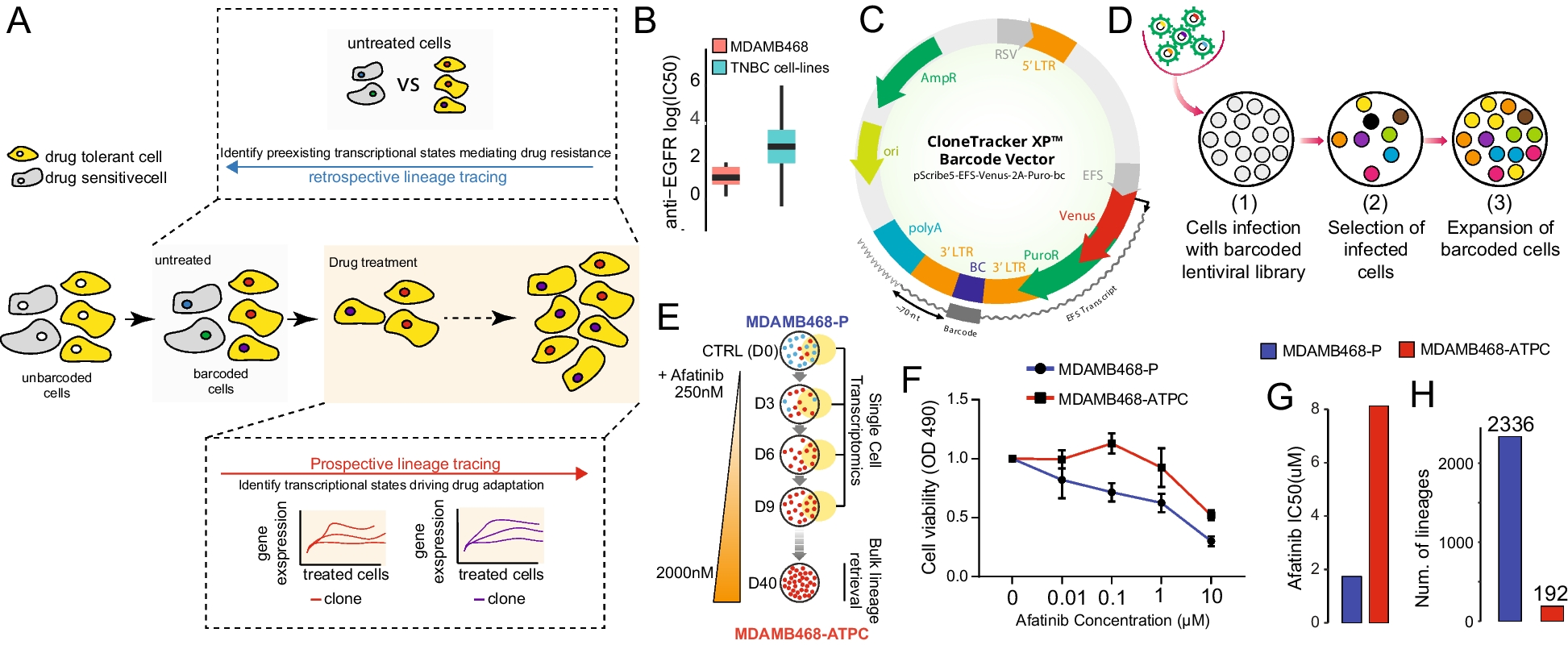
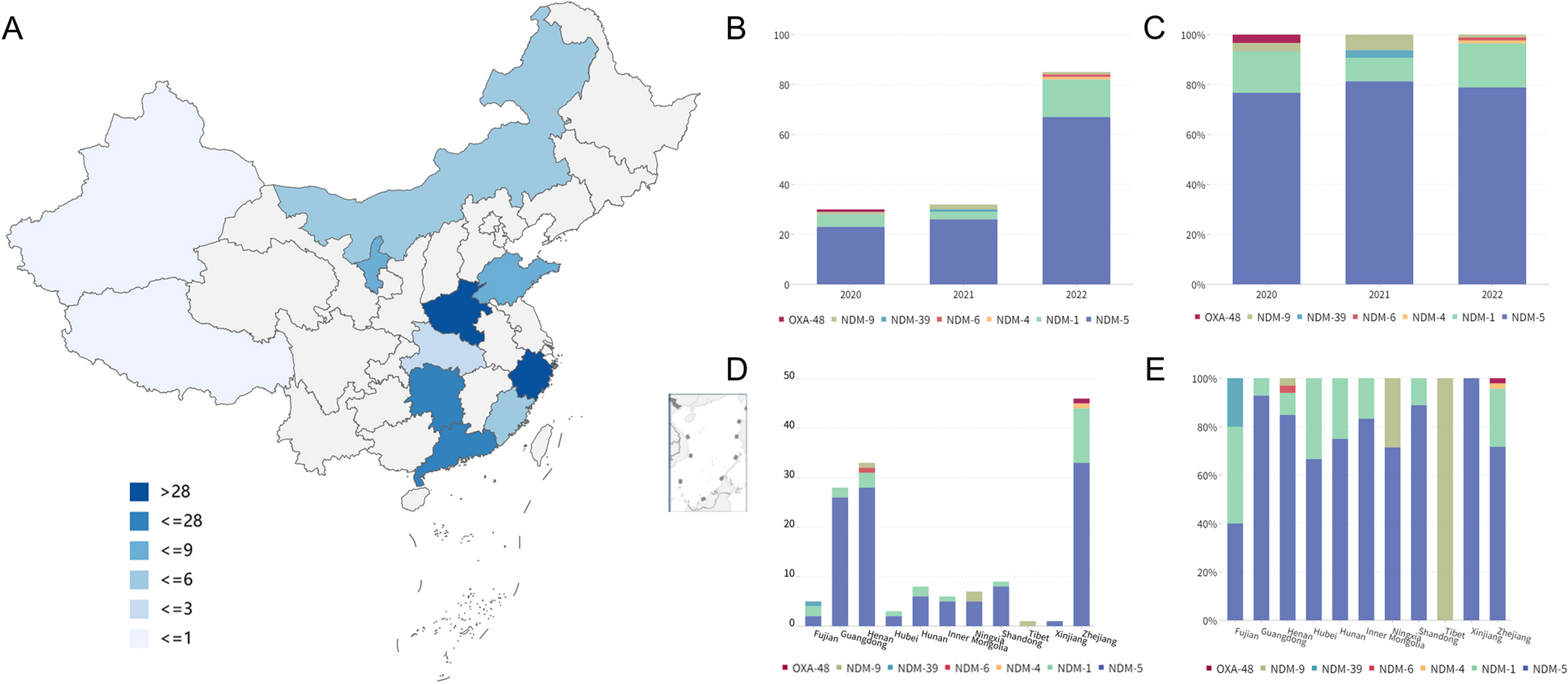
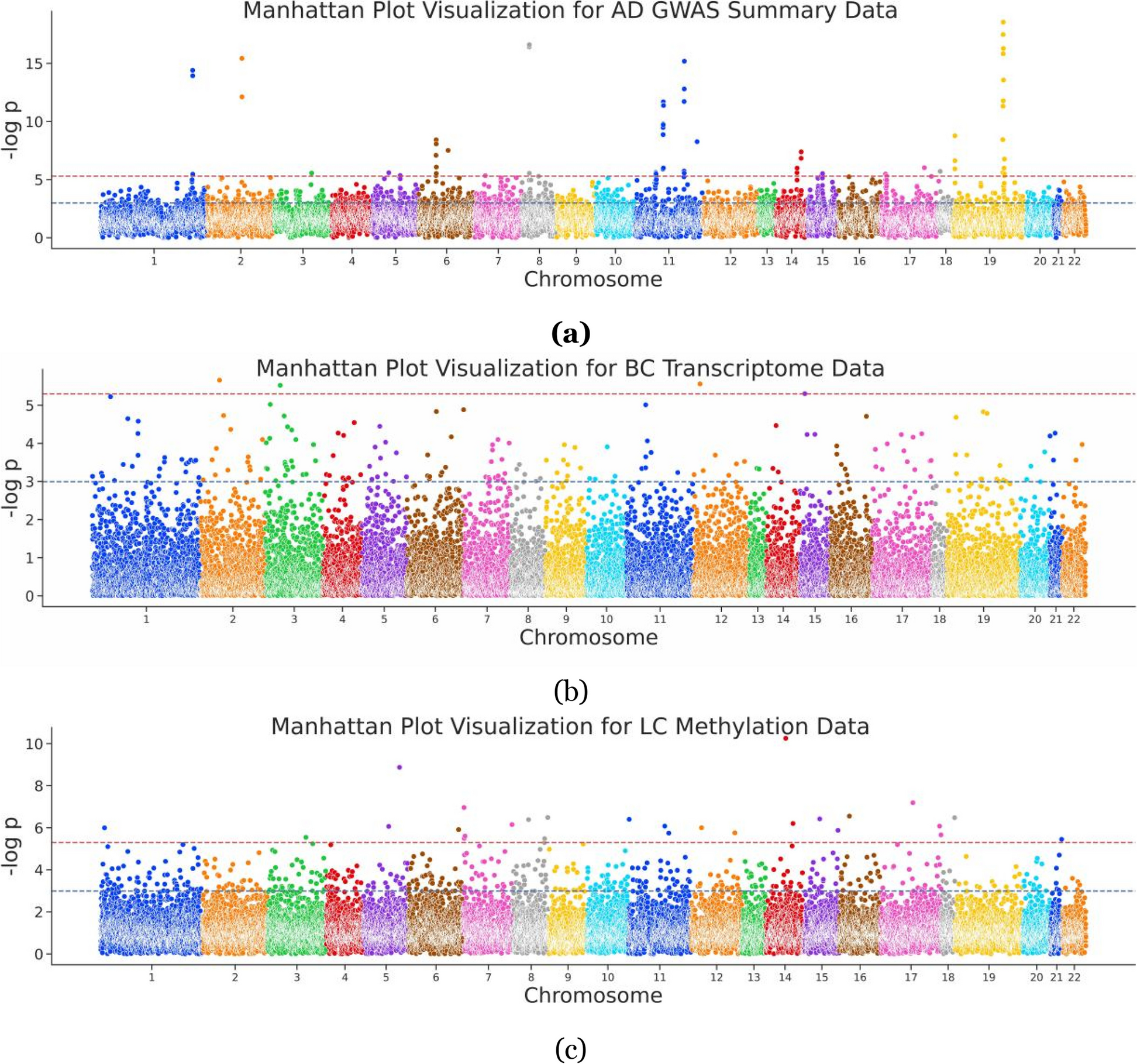
記住我





The mutation library consists of ~ 100 mitochondrion markers for speciation of P. falciparum, P. vivax, P. malariae, P. knowlesi and P. ovale ssp. (20 per species; Table 1), which also differentiate human from non-human affecting Plasmodium species. In brief, alignments of 75 mitochondrial genomes (51 human and 24 non-human Plasmodium; Fig. 1a) were used to construct a maximum likelihood phylogenetic tree. By annotating the tree branches with ancestral mutations [26], it was possible to define k-mers (31 bp) using kmc software [27], from which 20 SNPs exclusive to each human species were determined. Using the mitochondrial genome has the advantage of ~ 20 more copies than the nuclear genome in cells [8]. In addition, we included a set of established markers (n = 137) that differentiate geographical regions for P. falciparum (61; Eastern, Western and Horn of Africa, Southeast Asia, South America, Oceania), P. vivax (56; East Africa, South Asia, Southeast Asia, Southern Southeast Asia, South America) and P. knowlesi (20; Non-Borneo (Peninsular); Borneo – Macaca fascularis (Borneo-Mf), Borneo – Macaca nemestrina (Borneo-Mn)) [8, 17, 19, 20] (Table 2). In brief, these barcoding markers have been previously determined using the population differentiation FST statistic, and identifying scores of one, which indicate that the SNP allele is fixed in the region of interest and not present outside that location. Lastly, known drug resistance mutations (n = 37) across P. falciparum candidate genes [15] were also included in the library (Table 3) as well as genetic variants in putative drug-associated loci reported for other malaria species (e.g. orthologues of Pfcrt, Pfdhfr, Pfdhps, Pfkelch13 and Pfmdr1) [2, 18, 21]. The mutation libraries are available and hosted on the GitHub open-source site, with versioning capability (https://github.com/jodyphelan/malaria-db). Future changes in the species, geolocation and drug resistance mutation libraries can be discussed, tracked, and visualised as part of the GitHub hosting. This method of hosting also enables multiple users and developers across the malaria genomics community to contribute to the project.
Table 1 Predictions of Plasmodium species using Malaria-Profiler library (n = 9312)Fig. 1
Population structure of Plasmodium. a Circular maximum likelihood tree of 51 human and 24 non-human Plasmodium isolates using mitochondrial sequences shows perfect clustering of species as expected. This indicates the presence of a species-specific sequence which is exploited in the k-mer-based speciation function. Pf P. falciparum, Pv P. vivax, Pk P. knowlesi, Pcyn P. cynomolgi, Pm P. malariae, Poc P. ovale curtesi. b P. falciparum principal component analysis showing clustering by geographic region specifically separation between Southeast Asia and Oceania and Africa. c P. vivax principal component analysis showing clustering by geographic region. d P. knowlesi principal component analysis showing clustering by region (Peninsular (Pen-Pk) vs. Borneo Malaysia), and within Borneo based on host (Macaca fascularis (Mf-Pk) and Macaca nemestrina (Mn-Pk))
Table 2 Accuracy of the Malaria-Profiler library for geographical predictionsTable 3 Drug resistance based on known mutations in P. falciparum*In silico profilingThe Malaria-Profiler tool for the in silico analysis of species, geolocation and drug-resistant mutations was developed using the Python language (v3.8) with the pathogen-profiler library [12] and well-established bioinformatic tools such as trimmomatic [28], BWA [29] and SAMtools [30]. The pipeline can be customised (Additional file 1: Fig. S1), but in its default mode, reads are trimmed using trimmomatic (parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36) then mapped to the appropriate Plasmodium reference (e.g. P. falciparum 3D7, P. vivax PvPO1) using bwa (with default parameters). The raw data can be in an Illumina or ONT format (Additional file 1: Fig. S2). With the default settings, variants are called using freebayes [31] (parameters: -F 0.05) and annotated using snpEff [32] (parameters: -noLog -noStats), with the processing parallelised using GNU parallel [33]. Annotated variants are compared to the list of mutations in the Malaria-Profiler libraries. Variants can be filtered using coverage depth, allele frequency and per-strand depth parameters that can be set by the user. Additionally, other variant calling tools can be used instead of freebayes with bcftools [30] and gatk [34] also implemented. A minimum depth of tenfold coverage to call variants is set as the default (consistent with [5, 12]), but this can be changed by the user. Positions below this cut-off will be recorded and presented in the final report. The Malaria-Profiler pipeline calculates the proportion of the reads supporting each allele and reports this information, which can serve as a proxy for multi-infections. The Malaria-Profiler pipeline is available on GitHub (from https://github.com/jodyphelan/malaria-profiler) and can be installed through the bioconda channel [35]. Malaria-Profiler report outputs are written in json, txt and pdf formats, with options to collate data into multi-sample reports in a dashboard (Additional file 1: Fig. S2).
Sequencing data and variantsTo test the Malaria-Profiler tool, a dataset of 9321 strains was collated from Illumina WGS raw data in the public domain (see https://www.ebi.ac.uk/ena). This database includes P. falciparum (n = 7462; https://www.malariagen.net/apps/pf6 [36]; PRJEB2136, PRJEB2143, PRJEB4348, PRJEB4410, PRJEB4580, PRJEB4589, PRJEB4611, PRJEB4725, PRJEB5045, PRJNA108699 and PRJNA51255), P. vivax (n = 1661, https://www.malariagen.net/data/open-dataset-plasmodium-vivax-v4.0; PRJEB10888, PRJEB2136, PRJEB2140, PRJEB4409, PRJEB4410, PRJEB44419, PRJEB4580, PRJEB56411, PRJNA175266, PRJNA240366-240531, PRJNA271480, PRJNA284437, PRJNA295233, PRJNA420510, PRJNA432819, PRJNA603279, PRJNA643698, PRJNA65119, PRJNA655141, PRJNA67065, PRJNA67237, and PRJNA67239) [2, 21], P. knowlesi (n = 151; PRJEB10288, PRJEB1405, PRJEB23813, PRJEB28192, PRJEB33025, and PRJNA294104) [17, 20], P. malariae (n = 18; PRJEB33837) [18] and P. ovale (n = 5; PRJEB51041) [18]. Meta data, including the geographical site of sampling, was available from the same sources (e.g. www.malariagen.net/resources/open-data-resources). In addition, the mitochondrion reference genomes were obtained from GenBank for the neglected non-human malaria parasites (n = 24; e.g. P. cynomologi, P. inui, P. reichenowi and P. simiovale) were also included in the analysis. Alignments of mitochondrial genomes to the species library allow for the identification of primary Plasmodium infection and potential co-infections. Species were assigned if half of the 20 specific markers were identified in the data. For intra-species analysis, genome-wide SNPs and indels were called using established bioinformatic pipelines [2, 15, 17, 18]. In brief, the raw Illumina WGS data (fastQ format) were aligned to their respective reference genomes using BWA-mem software (default parameters). SNPs and short indels were called using the SAMtools and GATK software suites (see [19]). For ONT data, a similar pipeline was adopted, except sequence alignment was performed using minimap2 [37] software.
Using genomic data to inform on Plasmodium parasite speciation and geographical clusteringA maximum likelihood phylogenetic tree for Plasmodium species was constructed using RAxML-NG (v 0.9.0; 1000 bootstraps) software applied to mitochondrial genomes (n = 75; 5592 nucleotides), which were aligned using MUSCLE software [38] and filtered with the Gblocks tool [39] (default settings). The optimal substitution model of nucleotide or amino acid evolution for phylogenetic construction was determined by MEGAX software [40]. Parasite clustering within species (e.g. P. falciparum, P. vivax; total n = 9321), which is typically geographically based, was explored by performing a principal component analysis (PCA) on the isolates using pairwise Manhattan distances based on biallelic SNPs.
Malaria-Profiler performanceTo test the performance of the library, the WGS raw data for the 9321 strains were processed through the Malaria-Profiler pipeline to predict species, geolocation, and resistance status (for P. falciparum). The predictions were then compared to primary Plasmodium species and geographical recorded meta information (see www.malariagen.net/resources/open-data-resources), which were assumed to be the gold standard, and thereby allowed the calculation of the predictive accuracy of the Malaria-Profiler library. Phenotypic drug resistance status was not available for most isolates. Samples identified by Malaria-Profiler with potential co-infections were also analysed with Centrifuge software [41] to confirm the main Plasmodium species. When applying Centrifuge, the threshold for potential co-infection was based on the whole genome abundance (minimum 5%) and samples with > 1 Plasmodium species exceeding the threshold were assigned as mixed. To demonstrate the utility of WGS in the clinic, processed DNA (see [42] for protocols) from two isolates (isolate1, isolate2) sourced from two malaria patients at the Radboud University Medical Center were sequenced on the ONT MinION platform (v10) at The Applied Genomics Centre, LSHTM (accession numbers ERR11254081 and ERR11254083).
留言 (0)