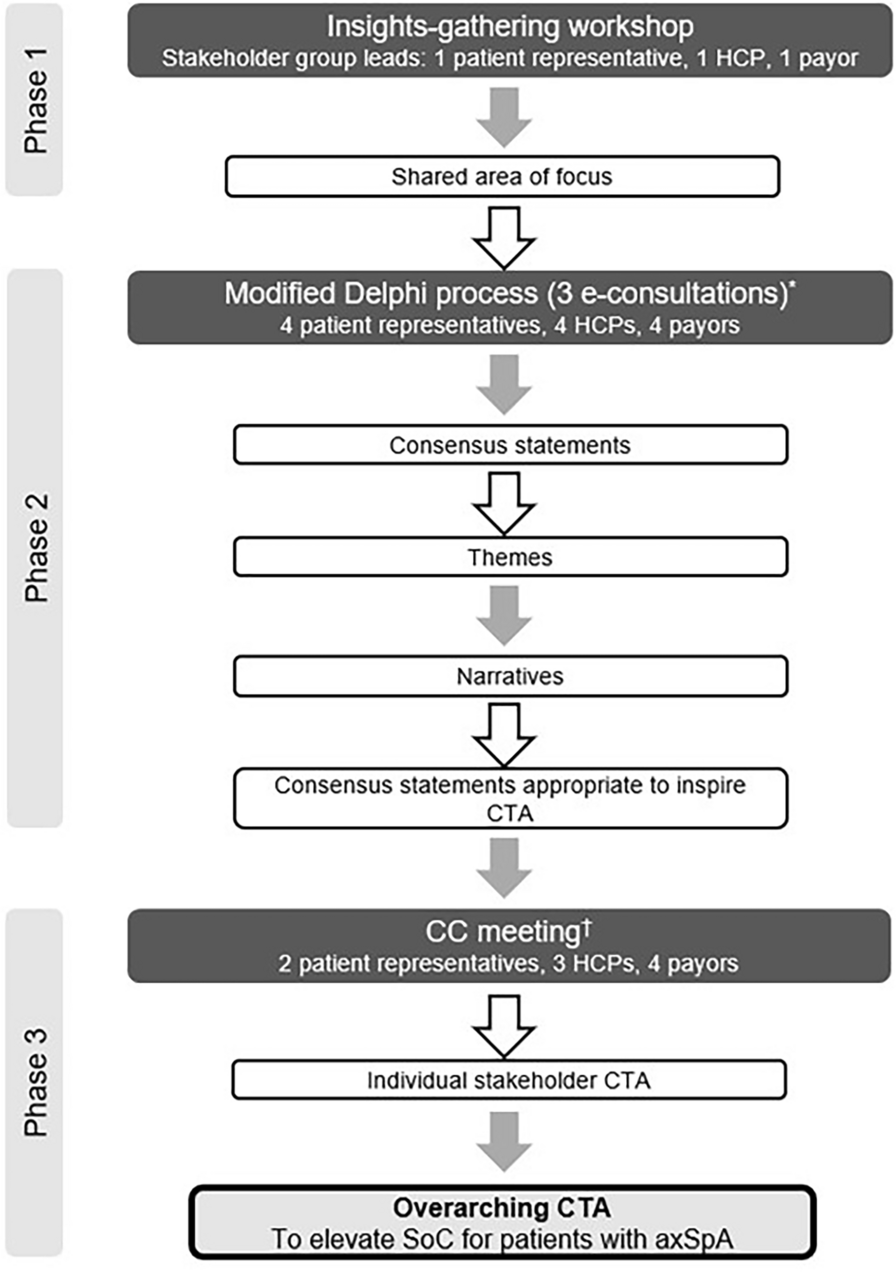
Study Design and Patients
The SPIRIT-P1 study design has been described previously [9]. Briefly, SPIRIT-P1 was a phase 3, multicenter, randomized, double-blind, clinical trial comparing the efficacy and safety of ixekizumab 80 mg every 4 weeks (Q4W) or every 2 weeks (Q2W) and an active reference arm of adalimumab (Humira®; AbbVie) 40 mg Q2W, with placebo in patients not previously treated with biologic agents for plaque psoriasis and PsA. All treatments were administered via subcutaneous injection, with patients randomized to the ixekizumab treatment group receiving a starting dose of 160 mg. Patients with an inadequate response at week 16, regardless of their treatment group, were required to add or modify concurrent treatment, with investigators/study personnel/patients blinded to the inadequate response criteria. Inadequate responders remained on their original dose of ixekizumab, or if receiving adalimumab or placebo, were re-randomized to ixekizumab Q4W or Q2W in a 1:1 ratio.
Enrolled patients were adults (≥ 18 years) who fulfilled the Classification Criteria for Psoriatic Arthritis, had active PsA defined as the presence of ≥ 3 of 68 tender joints and ≥ 3 of 66 swollen joints, had either ≥ 1 PsA-related hand or foot joint erosion on centrally read X-rays or C-reactive protein (CRP) > 6 mg/l, and active psoriatic skin lesions (plaque) or a documented history of plaque psoriasis. Patients with symptoms consistent with axial involvement were not excluded from the SPIRIT-P1 study.
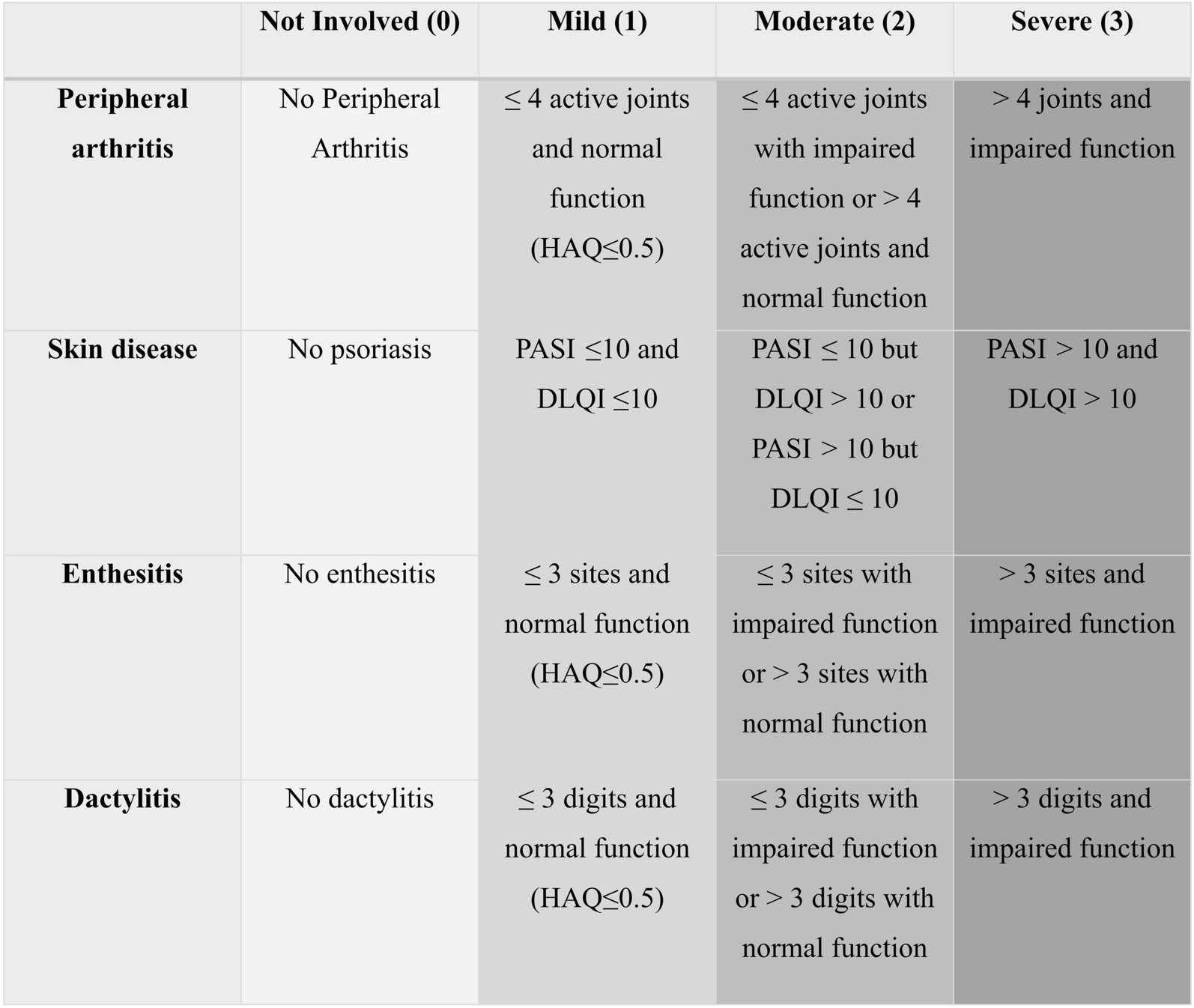
As PsA can affect various domains in addition to peripheral arthritis, we defined PsA severity by considering multiple clinical domains. The total mCPDAI score is a composite measure defining the severity of individual domains including peripheral arthritis, skin, enthesitis, dactylitis, and axial manifestation, but not including Ankylosing Spondylitis Quality of Life (ASQoL). It has previously been reported that the cutoff for high disease activity for CPDAI is ‘8’ [18]. In SPIRIT-P1, mCPDAI (excluding ASQoL) was used as one of the secondary endpoints. Taken together, for this subgroup analysis of SPIRIT-P1, we defined the severe population using both the total and peripheral arthritis scores. Hence, patients with a mCPDAI total score > 7 [16] and peripheral arthritis score = 3 (the highest score) were defined as having severe symptoms.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committees at all sites where these studies were conducted and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study, and all participants consented to the publication of study results. Protocols and consent forms were approved by the institutional review board or ethics committee of each site, including the Western Institutional Review Board (SPIRIT-P1). A listing of individual sites for SPIRIT-P1 is included in the supplement of the primary manuscript [9]. SPIRIT-P1 was registered at ClinicalTrials.gov, NCT01695239.
Study Assessments
Efficacy was measured by the proportion of patients achieving ACR 20, ACR 50, and ACR 70 responses from week 0 to week 24; change from baseline in ACR core set at weeks 1, 12, and 24; the proportion of patients with change from baseline in van der Heijde modified total Sharp score (mTSS) from week 0 to week 24; the proportion of structural damage progression, cutoffs ≤ 0, ≤ 0.5, and ≤ 0.95 (the smallest detectable change [9]), at week 24; the proportion of patients achieving PASI 75, PASI 90, and PASI 100 at week 24; the proportion of patients achieving disease activity index for psoriatic arthritis (DAPSA) low disease activity (LDA) (> 4 and ≤ 14) and DAPSA remission (≤ 4) from week 0 to 24; and change from baseline in 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) from week 0 to 24.
Safety was assessed by the proportion of patients experiencing treatment-emergent adverse events (TEAEs), serious adverse events, and study discontinuation due to adverse events (AEs). AEs of special interest included cytopenia, infection, serious infections, injection site reactions, hepatic events, allergic reaction/hypersensitivity, depression, and malignancy.
Statistical Analyses
Statistical analyses have been previously described for the overall population [9]. Efficacy assessments were conducted on the overall intent-to-treat population (all randomized patients), the severe intent-to-treat population (defined as mCPDAI total score > 7 and peripheral arthritis score = 3), and the non-severe intent-to-treat population. All efficacy endpoints were assessed at a significance level of p < 0.05 with no adjustment for multiplicity.
Post hoc subgroup analyses of categorical endpoints for the severe and non-severe populations were based on a logistic regression analysis with treatment, subgroup (severe/non-severe), and treatment-by-subgroup interaction (tested at the 10% significance level). Patients were considered non-responders (non-responder imputation, NRI) if they did not meet the clinical response criteria for categorical responses or were missing categorical response data at a timepoint of interest. Patients who were eligible for rescue therapy at week 16 were analyzed as non-responders after week 16. Post hoc subgroup analyses of continuous variables for the severe and non-severe populations were based on an analysis of covariance model including treatment, subgroup, treatment-by-subgroup (tested at the 10% significance level), and baseline value of the response variable.
With the exception of analyses for mTSS, a last-observation-carried-forward model was used for all continuous efficacy endpoints. For patients who discontinued study treatment for any reason, the last non-missing observation before discontinuation was carried forward to the corresponding endpoint for evaluation. However, for patients eligible for rescue therapy at week 16, the last non-missing observation up to week 16 was carried forward to the corresponding endpoint for evaluation. Randomized patients without at least one post-baseline observation were not evaluated.
The linear extrapolation method was used for the analyses of mTSS. For patients who discontinued the study or the study treatment, or missed a radiograph for any reason, baseline data and the most recent radiographic data were used for linear extrapolation to impute missing data at subsequent scheduled timepoints. For patients who commenced rescue therapy at week 16, or at any timepoint thereafter, baseline data and the most recent post-baseline radiographic data up to week 16, adjusted for time, were used for linear extrapolation.
Safety analyses were conducted on the overall safety population (all patients who took at least one dose of study medication), the severe safety population (defined as mCPDAI total score > 7 and peripheral arthritis score = 3), and the non-severe safety population using descriptive statistics.
The adalimumab 40 mg Q2W treatment arm served as active reference for comparison with placebo. The study was not powered to test equivalence or non-inferiority of ixekizumab vs. adalimumab. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).



留言 (0)