This article provides clinical and genetic data on 18 individuals from 17 Turkish families and 1 Syrian family with disease-causing mutations in COL4A3, COL4A4, and COL4A5 and coinherited genetic findings.
AS is inherited as X-linked (XLAS), autosomal recessive (ARAS), and autosomal dominant (ADAS). AS is characterized by impaired synthesis and deposition of collagen IV alpha3/4/5 networks in the BMs of the glomerulus, cochlea, and eye [7]. Hematuria is the most common symptom of AS, and males are affected more often than women [14]. Interestingly, we had more female patients, and COL4A4 was the most common mutated gene in our study, which differs from the literature [9].
The clinical manifestations of ADAS patients vary from asymptomatic to renal failure, with no association with the causative gene or variant type. Due to the many phenotypes associated with ADAS, it is underdiagnosed in clinical practice [5, 15]. Few individuals with heterozygous COL4A3 or COL4A4 mutations develop ESRD, hearing loss, or a lamellated GBM. Hence, there is no consensus regarding the term ADAS. Nonetheless, the prognosis of patients with ADAS is not always favorable, and a small but unexpected proportion of them suffer renal failure. Given that most other family members with similar mutations maintain normal renal function throughout their lifetimes, there is no obvious explanation for this. This discrepancy demonstrates that there are other causes of renal failure for these patients [16]. Patients with ADAS have a slower rate of progression to ESRD and are less likely to have extrarenal symptoms than patients with XLAS. ESRD and SNHL often manifest in later adulthood, and ocular involvement is infrequent [2, 3].
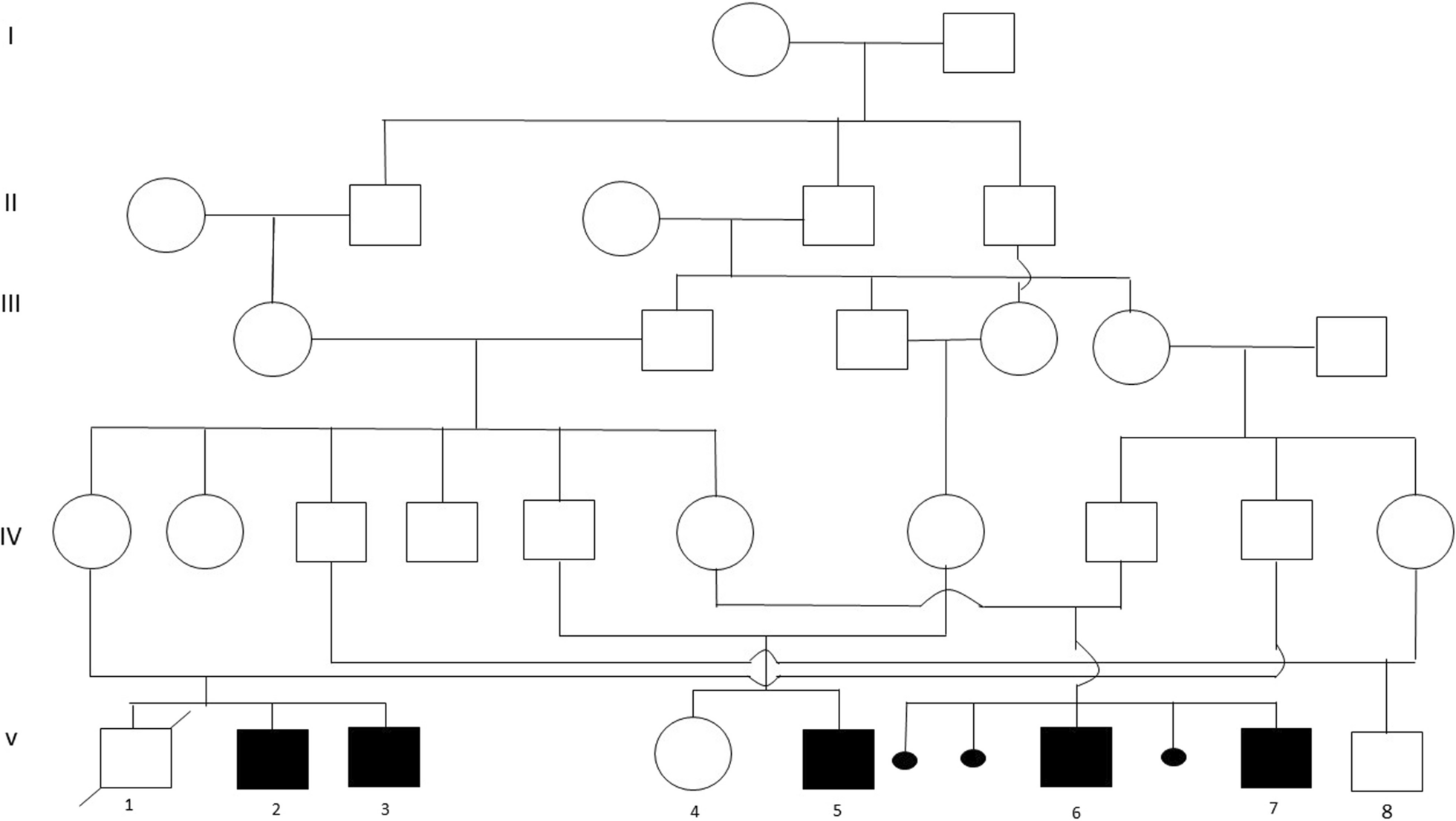
Fallerini et al. [13] and Mencarelli et al. [12] proposed digenic inheritance in AS, which could explain the variable expressivity of AS. Patients with heterozygous mutations in two distinct collagen IV genes suffer renal failure later than those with XLAS or ARAS but earlier than those with ADAS [12]. In patient 10, both heterozygous likely pathogenic “COL4A3, c.3536del, p.Pro1179Glnfs*42” and “COL4A5, c.3190_3192dup, p.Asp1064dup” mutations were detected. Her mother had “COL4A5, c.3190_3192dup, p.Asp1064dup” mutation, and her father had the “COL4A3, c.3536del, p.Pro1179Glnfs*42” mutation. The patient had proteinuria, hematuria, focal segmental glomerulosclerosis (FSGS), interstitial fibrosis, tubular atrophy, and hypertension. The COL4A3/COL4A5 combination has been reported with different mutations [13]. This case provides more evidence of digenic inheritance in AS.
Modifiers play an essential role in AS. The NPHS2, LAMA5, LAMB2, APO1E, CFHR5, ACTN4, PODXL, WT1, TRPC6, CD2AP, and INF2 genes have been reported to be modifiers [16]. In our study, coinherited genetic mutations were detected in six genes, including TTC21B (responsible for Nephronophthisis 12 (MIM: 613820) autosomal dominant and recessive and Short-rib thoracic dysplasia 4 with or without polydactyly (MIM: 613819), autosomal recessive), SLC34A1 (responsible for Fanconi renotubular syndrome 2 (MIM: 613388), autosomal recessive, hypercalcemia, infantile, 2 (MIM: 616963), autosomal recessive, nephrolithiasis/osteoporosis, hypophosphatemic, 1 (MIM: 612286), autosomal dominant), PKHD1 (responsible from polycystic kidney disease 4, with or without hepatic disease (MIM: 263200), autosomal recessive), FANCC (responsible for Fanconi anemia, complementation group C (MIM: 227645), autosomal recessive), and MEFV (responsible for familial Mediterranean fever (MIM: 134610, 249100), autosomal recessive and dominant), GJB2 (responsible for Deafness (MIM: 601544, 220290), autosomal dominant and recessive).
In Patient 1, since the COL4A4 mutation was heterozygous, the likely pathogenic “TTC21B, c.2599C > T, p.Arg867Cys” mutation may be responsible for the early onset of clinical symptoms and renal failure both in herself and her mother. Patient 4 was only three years old. At very early ages, hematuria with a heterozygous COL4A3 mutation is not expected. Likely pathogenic “SLC34A1, c.558dupC, p.Ile187Hisfs*26/c.272_292del, p.Val91_Ala97del” and pathogenic “PKHD1, c.107C > T, p.Thr36Met/c.5513A > G, p.Tyr1838Cys” mutations could be the reason patient 4 suffered from bilateral renal cysts and aggravated hematuria. Patient 14 had heterozygous splicing COL4A4 mutation and some dysmorphic features. Her mother had the same COL4A4 mutation and had no clinical symptoms. Pathogenic homozygous “FANCC, c.456 + 4A > T” mutation could explain not only the dysmorphic features but also the finding of AS. Patient 15 had renal failure at a very early age. Homozygous MEFV M694V mutation along with a frameshift homozygous COL4A3 were detected. MEFV mutation could explain the patient’s fever, abdominal pain attacks, aggravated renal failure, and AS. Although parents were carriers of both mutations, they had no clinical findings.
Patient 16 had hematuria starting at age six and bilateral sensorineural hearing loss. She had a heterozygous missense COL4A4 mutation, which replaced glycine with arginine. It has been proposed that mutations cause different degrees of pathogenicity: more severe mutations, such as frame deletions, stop codons, and those resulting in chain termination, result in more severe phenotypes, for example, worse renal function, along with increased hearing and vision alterations. In contrast, missense mutations with glycine replacement are related to a less aggressive form of the disease [2]. The “GJB2, c.35del, p.Gly12fs” homozygous pathogenic variant led to bilateral severe sensorineural hearing loss and clinical signs at an earlier age, which were more severe than expected. The parents had no clinical symptoms.
There is still some confusion about AS, including phenotypic variability. Patients with the same mutations showed different features, from mild to severe. Some of our patients had a more severe phenotype compared to their parents or siblings, although they had the same mutation. Clinical variability may be attributed to several factors, such as different expressivity, incomplete penetrance, and the impact of mutant alleles on wild-type proteins. Other genes may be involved in the phenotype variability, acting as disease modifiers. The high familial variability suggests that genetic modifiers, as well as epigenetic or environmental variables, may play a role. Therefore, comprehensive sequencing analysis by NGS should be the primary strategy for gene screening in AS.
Another issue is that it is not easy to distinguish ADAS from ARAS. Some mutations exhibit a dominant and recessive inheritance pattern, complicating our understanding of the genotype–phenotype correlation and mode of inheritance. This was also evident in our patients. Even after testing healthy parents and siblings, it was not easy to diagnose ADAS or ARAS based on the clinical findings. Patients with ADAS exhibit a broad range of clinical manifestations, from asymptomatic to renal failure, with no evident correlation to the causal gene or variant type. Due to the diverse phenotypes associated with ADAS, it is underdiagnosed in clinical practice [15, 17]. Establishing the mode of inheritance is critical for genetic counseling, identifying other at-risk family members, assessing the status of potential kidney donors, and for prenatal and preimplantation genetic diagnosis. A proper ADAS diagnosis will lead to genetic counseling for families, avoidance of kidney biopsies, and more effective treatment strategies.
The third problem is the difficulty in predicting the phenotype and prognosis based on mutations. Patients with non-functional early truncating mutations would be expected to have severe clinical manifestations, whereas missense mutations with glycine substitution are associated with a less aggressive form of the illness [2]. According to this presumption, most of our patients should have had a severe phenotype; however, they had milder phenotypes. The relationship between genetic abnormalities, disease etiology, and clinical findings remains unknown. Founder and recurrent mutations are not frequent in AS, making it harder to predict the effect of the mutations.






Comments (0)