
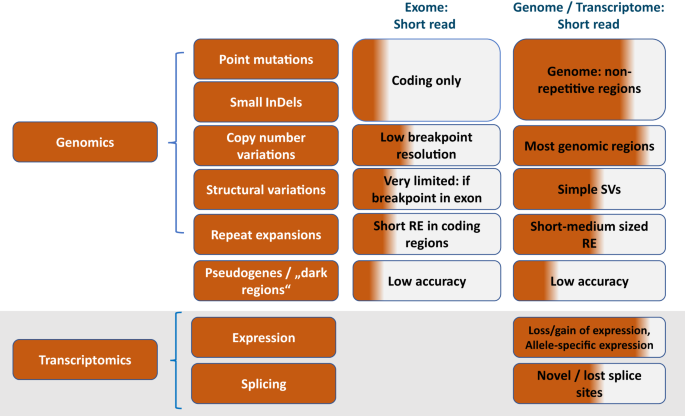
Remember me

The burden of in silico predicted, splice-disrupting variants in definitively associated and phenotypically concordant disease genes was evaluated in 1242 unrelated participants with inherited heart disease or SUD. Hypertrophic cardiomyopathy (HCM) was the most common diagnosis in the cohort (n = 720), followed by SUD (n = 203), dilated cardiomyopathy (DCM) (n = 143), Brugada syndrome (BrS) (n = 66), long QT syndrome (LQTS) (n = 55), arrhythmogenic cardiomyopathy (ACM) (n = 34) and catecholaminergic polymorphic ventricular tachycardia (CPVT) (n = 21). We found 88 rare in silico predicted, splice-disrupting variants in 128 out of 1242 (10.3%) participants, and only nine participants had an alternative genetic cause for their disease (Supplementary Table 1). Within each disease group studied, approximately 10% of participants carried an in silico-predicted splice-disrupting variant, except for BrS, where only 1 out of 66 participants (1.5%) did (Fig. 1). The genes with the most splice-disrupting variants were MYBPC3 (36/88, 41%), followed by TTN (14/88, 16%), FLNC (6/88, 7%), MYH7 (6/88, 7%) and KCNQ1 (5/88, 6%), with few variants, or none, in the remaining genes (Fig. 2). Self-reported ethnicities were available for 122 participants with splice-disrupting variants, with 95 European, 12 Asian, 6 North African, 8 Oceanian, and 1 ‘Peoples of the Americas’ (Supplementary Table 1).
Fig. 1: The proportion of participants with an in silico-predicted splice-disrupting variant.
Total participant counts within each disease group are shown in the boxes above each bar plot. The overall average of participants with an in silico-predicted splice-disrupting variant (10.3%) is highlighted by the red dotted line. HCM hypertrophic cardiomyopathy, SUD sudden unexplained death, DCM dilated cardiomyopathy, BrS Brugada syndrome, LQTS long QT syndrome, ACM arrhythmogenic cardiomyopathy, and CPVT catecholaminergic polymorphic ventricular tachycardia.
Fig. 2: The percentage of unique in silico-predicted splice-disrupting variants in each gene.
Percentage of unique in silico-predicted splice-disrupting variants found in a list of 32 genes prioritised based on phenotypically concordant genes established by the ClinGen Curation Expert groups for hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic cardiomyopathy, long QT syndrome, Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia.
We compared the burden of rare putative splice-disrupting variants in our disease cohorts with gnomAD (v2.1.1) exomes control populations. There was a significant excess of splice-disrupting variants in PKP2 in people with ACM (excess burden in cases = 5.9%, P < 0.001), FLNC (2.7%, P < 0.001) and TTN (2.8%, P < 0.001) in people with DCM, MYBPC3 (8.2%, P < 0.001) and MYH7 (1.3%, P < 0.001) in people with HCM, and KCNQ1 in people with LQTS (3.6%, P < 0.001) (Supplementary Table 2). Statistical significance of these six genes was maintained when repeating the burden test with only European cases and European Non-Finnish gnomAD controls (Supplementary Table 3). The excess of putative splice-disrupting MYH7 variants is primarily driven by six HCM participants having a Glu849Gly missense change due to a c.2681 A > G variant at the second nucleotide of exon 23, which was predicted to disrupt splicing by the in silico tools, adapting boosting (ADA) and random forests (RF)12.
Mapping the location of splice-disrupting variantsThe location of in silico predicted, splice-disrupting variants within the donor and acceptor sites was assessed (Fig. 3, Supplementary Table 4). Twenty-four variants (27%) were in the donor site, of which 13 variants disrupted the essential ‘GT’ dinucleotide and three clustered at the +5 intronic nucleotide position. Twenty-four variants (27%) were in the acceptor site, of which 14 disrupted the essential ‘AG’ dinucleotide. Ten variants (11%) in the first and last nucleotide of an exon previously annotated as missense changes were predicted to disrupt the adjacent splice site.
Fig. 3: Location of unique in silico-predicted splice-disrupting variants in the essential splice site region.
The positions of the essential GT/AG dinucleotides are shown in red. The splice donor site consists of the last three nucleotides of the exon and the first six nucleotides of the intron. The acceptor site consists of the last 20 nucleotides of the intron and the first three nucleotides of the exon. The intervening intron is truncated ‘//’. Three small deletions within the splice site region are not shown.
Outside of the canonical splice sites, there were 25 variants in deep intronic regions predicted to create new donor (n = 15) or acceptor (n = 10) sites, and 15 exonic variants annotated as missense, nonsense, or synonymous variant, predicted to create new donor (n = 11) or acceptor (n = 4) sites (Fig. 4, Supplementary Table 4).
Fig. 4: Classification of in silico-predicted splice-disrupting variants.
Variants located in the −3 to +6 region of the donor site or the −20 to +3 region of the acceptor site are classified as ‘Splice site region’ variants. All other intronic variants were labelled as ‘deep intronic region’. Remaining variants in the exons, including missense, nonsense, and synonymous variants, were categorised as ‘exonic region’.
RNA sources for amplification of definitively disease-associated cardiac genesWe determined which sources of mRNA would support RT-PCR amplification of the definitively associated inherited heart disease genes. In total, 21 out of 31 (68%) genes were amplified concordantly using mRNA extracted from blood, induced pluripotent stem cell-derived cardiomyocytes and myectomy tissue (Supplementary Table 5, Supplementary Fig. 2). Nine genes only amplified in induced pluripotent stem cell-derived cardiomyocytes and myectomy tissue, (CASQ2, TECRL, FLNC, MYH7, TNNT2, ACTC1, MYL2, MYL3 and TNNI3), while TRDN only amplified in myectomy tissue. Seven genes amplified more than one product due to alternative splicing (RYR2, TECRL, BAG3, MYH7, ACTC1, TNNI3 and TPM1) (Supplementary Fig. 2). PLN, which is definitively associated with DCM, was not included as the single coding exon in the MANE transcript, NM_002667.5, does not undergo splicing.
Functional studies of in silico-predicted splice-disrupting variantsSix in silico predicted, splice-disrupting variants without prior mRNA testing were functionally studied using blood RNA from the affected individuals and family members who carried the variant, where available (Table 1). We assessed three variants in donor splice sites. Amplification of mRNA from a female with LQTS and a KCNQ1 c.477+5 G > A variant revealed exon 2 skipping resulting in a 91 bp deletion, leading to a frameshift and premature termination codon (Supplementary Fig. 3a). An RYR2 c.848+1 G > A variant in a female diagnosed with familial CPVT caused exon 11 skipping, leading to an in-frame deletion of 25 amino acids (Supplementary Fig. 3b). A TTN c. 63793 G > A missense variant at the last nucleotide of exon 307 was found in a male with SUD, his uncle with DCM, and his clinically unaffected father, totalling six meiosis. RNA extracted from the father showed the retention of intron 307, leading to a frameshift and a premature stop codon (Supplementary Fig. 3c). We assessed two variants in acceptor splice sites. A missense variant in KCNQ1 c.781 G > A, found in a female with LQTS, caused the skipping of exon 6, leading to an in-frame deletion of 47 amino acids (Supplementary Fig. 3d). An MYBPC3 c.1458-7 C > A variant found in a male with HCM caused a 5 bp extension of exon 17, leading to a premature stop (Supplementary Fig. 3e). Finally, a deep intronic c.1224-80 G > A variant in MYBPC3 found in a male with HCM created a new splice acceptor site resulting in a 78 bp extension of exon 14, leading to an in-frame inclusion of 26 amino acids (Supplementary Fig. 3f).
Table 1 Functional study outcomes and sequence variant classification.Classification of sequence variantsWe classified the pathogenicity of all 88 in silico predicted, splice-disrupting variants; 43 were classified as pathogenic or likely pathogenic and 45 as VUS (Fig. 4, Supplementary Table 4). Most variants in the splice site regions were pathogenic or likely pathogenic, whereas most variants outside these regions were VUS. The results of our RNA-based functional studies, and previously published functional studies, were available for 29 out of 88 variants (33%). They confirmed that 19 variants caused a frameshift leading to a premature termination codon, six caused an in-frame insertion or deletion in the mRNA, one disrupted splicing with unreported consequences, one did not alter splicing, and one resulted in impaired protein function. One variant showed inconsistent results across multiple studies. These mRNA studies supported the reclassification of 11 VUS as likely pathogenic, and 3 likely pathogenic variants were upgraded to pathogenic (Supplementary Table 4). The reclassification of 11 VUS as likely pathogenic allowed these variants to be used for cascade genetic testing. Of these 11 VUS, six variants co-segregated with a concordant phenotype in 10 family members, one heterozygous family member with an unknown clinical status, and one family member who was genotype positive; phenotype negative.
Comments (0)