
記住我







All animal experiments were performed with the approval of the Garvan Institute and St. Vincent’s Hospital Animal Ethics Committee (protocol IDs AEC 08/20, 11/51, 14/40 and 17/28) as well as the animal ethics committee of the Florey Institute of Neuroscience and Mental Health (for ECoG recordings; protocol ID AEC 14-025) and the animal ethics committee of Western Sydney University (for tissue collection from 5xFAD mice; protocol ID A13397), in accordance with National Health and Medical Research Council animal experimentation guidelines and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (2013). Animals used in electrophysiology experiments were performed with the approval of the University of Otago Animal Ethics Committee (ET10-15). For all studies, only male mice were used. Mice were housed in individually ventilated cages (IVC) and maintained on a 12 h light/dark cycle (lights on at 7:00am).
5xFAD miceNote: these mice were only used for Sup Fig. 5b. Hemizygous and non-transgenic littermates were from the B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax (aka: 5xFAD) line (MMRRC stock # 34840). Transgenic mice express human amyloid precursor protein (hAPP) containing the Swedish, Florida and London familial Alzheimer’s disease mutations as well as the Presenilin 1 (PS1) gene with two mutations. Both genes are regulated by the mouse Thy1 promoter [66]. Mice were maintained on a mixed background by breeding within the colony and were housed at a maximum of three per cage. Genotyping was performed through PCR amplification of genomic DNA using standard practices and using primer sequences recommended by The Jackson Laboratory.
J20 miceHemizygous transgenic and non-transgenic littermates were from the B6.Cg-Tg(PDGFB-APPSwInd)20Lms/2J (aka: J20) line (MMRRC stock #34836), which express hAPP containing both the Swedish and Indiana mutations, under a PDGF-β promoter [67]. Mice were maintained on a C57BL/6J background and were housed at a maximum of five per cage. Genotyping was performed through PCR amplification of genomic DNA using standard practices and using primer sequences recommended by The Jackson Laboratory.
Gria2 tm1BViss miceTo develop the Gria2tm1BViss mice (i.e. exonically encoded GluA2(R)), a construct was generated from DNA cloned from a 129 SvEv DNA genomic library (Sup Fig. 1A). A neomycin gene, flanked by loxP sites, was placed downstream of exon 11. In addition, a single point adenosine to guanine mutation was made at the Q/R editing site of intron 11 (CAG ➔ CGG). The construct was electroporated into CCE embryonic stem (ES) cells, which were derived from 129SvEv mice. Colonies resistant to G418 were isolated and an ES cell colony that contained the allele was identified. This ES cell colony was electroporated with Cre-expressing plasmid and re-plated in the absence of G418, thus excising the neomycin and leaving a single loxP site in addition to the point mutation. Genotyping was performed through PCR amplification of genomic DNA using standard practices. Specific oligonucleotide primers for the Gria2 wild-type allele were: common forward- 5′-GTG TCT CTT GGG GAA GTT CAA T-3′; and reverse- 5′- TGA TAT ATT TCC CTC TTC TCA GCC AGT GG -3′. For the targeted allele, a reverse primer was designed from within the loxP sequence as follows: reverse- 5′-TGC CCA CAT CTA AGA TTG TTG GAC-3′. Additionally, presence of only the targeted Gria2G/G transcript (aka: edited GluA2(R)) was confirmed via RT-PCR of the Gria2 gene containing the Q/R site and subsequent digestion with the BbvI restriction enzyme as previously described [58].
Gria2 tm1BViss x J20 crossGria2tm1BViss mice were maintained on a C57BL/6J background for at least 10 generations before being crossed with hemizygous J20 mice. To obtain littermates of all possible genotypes, the bi-transgenic colony was maintained by mating female hemizygous Gria2tm1BViss mice with double hemizygotes from the cross colony. Of the resulting possible genotypes, four were assessed (see Table 1 below). Mice were housed at a maximum of five per cage. For behavioral studies that involved the radial arm maze (RAM), mice were housed individually. Food and water were available ad libitum unless dietary restrictions were required for mice undergoing RAM testing.
Table 1 Description of genotypes used in study. All mice are littermates from crossing Gria2tm1BViss mice with J20 miceElectrophysiologyField potential electrophysiologyAll recordings were made by an experimenter blind to the genotype of each mouse. Mice (33–54 weeks of age), were deeply anaesthetised with ketamine (100 mg/kg, i.p.), and the brains removed and chilled in ice-cold and oxygenated modified artificial cerebrospinal fluid (aCSF) in which sucrose was substituted for NaCl (composition in mM: sucrose 210, glucose 20, KCl 2.5, NaH2PO4 1.25, NaHCO3 26, CaCl2 0.5, MgCl2 3, pH 7.4 when gassed with 95% O2-5% CO2). Whole hemisphere parasagittal slices (400 µm) containing transverse sections of the dorsal hippocampus were cut in a manner similar to that described previously [68] using a vibratome (VT1000, Leica Microsystems). Slices were transferred to a custom-built incubation chamber containing aCSF (mM: 124 NaCl, 3.2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 10 D-glucose) bubbled with carbogen. Slices were incubated at interface for 30 min at 32 °C and then at room temperature for at least 90 min. After this recovery period, they were transferred to a recording chamber at 32.5 °C and superfused (2 mL/min) with oxygenated aCSF.
Baseline field excitatory postsynaptic potentials (fEPSPs) in area CA1 of the hippocampus were elicited by stimulation of the Schaffer collateral-commissural pathway at 0.017 Hz (diphasic pulses, 0.1 ms half-wave duration) using a Teflon-coated 50 μm tungsten wire monopolar electrode (A-M Systems Inc). Evoked responses were recorded using a glass microelectrode filled with aCSF (2–3 MΩ) and placed in stratum radiatum. During periods of baseline recording, the stimulation intensity was adjusted to elicit a fEPSP with an initial slope value of 40% of the maximum slope elicited when delivering 200 µA of current. Drugs were bath-applied by switching to an identical preheated and oxygenated aCSF solution that contained the compound of interest.
Once fEPSPs were established, we conducted three electrophysiological procedures in stratum radiatum. These were: paired-pulse facilitation (PPF) to assess presynaptic short-term plasticity; an input-output test to assess basal synaptic transmission; and induction of long-term potentiation (LTP) to assess synaptic plasticity. PPF was induced in stratum radiatum by delivering paired stimuli (3 pairs at 10 s intervals, 1 mV initial response) at interpulse intervals ranging from 20–200 ms in the presence of D-AP5 (50 µM, Tocris). PPF was expressed as a ratio of the response amplitudes and was calculated as EPSP 2 amplitude/EPSP 1 amplitude. Basal synaptic transmission was determined across stimuli of increasing intensity ranging from 10–200 µA (average slope of 3 responses at each stimulus intensity) to generate an input-output (I-O) curve. LTP was induced by applying two trains of standard theta-burst stimulation (TBS, each train containing 10 bursts of 5 pulses at 100 Hz delivered at 200 ms intervals, 30 s between each train) at baseline stimulus intensity. Baseline pulses were delivered every 30 s for 20 min prior to the TBS and for 60 min after. Baseline recording continued for a further 60 min. The initial slopes of the fEPSPs were measured, and each response expressed as a percentage change from baseline, defined as the average of the last 30 responses before TBS delivery. Initial LTP magnitude was calculated as the average slope of the first 20 responses after TBS, while the final LTP magnitude was calculated as the average slope of the last 20 responses, all normalised to the baseline.
Patch-clamp electrophysiologySlice preparation was as for the field recording experiments except that animals were anaesthetised with pentobarbital (200 mg/kg; i.p.) and transverse whole hemisphere slices (400 μm) containing the dorsal hippocampus were cut. Hippocampal CA1 whole-cell recordings were performed in mice at 43–54 weeks of age. The Schaffer collateral pathway in stratum radiatum was stimulated using 50 μm Teflon-insulated tungsten monopolar electrodes and evoked excitatory postsynaptic currents (EPSCs) were recorded from visualised CA1 pyramidal cells using a glass patch electrode at holding voltages of -70 mV, -60 mV, -40 mV, -20 mV, 0 mV, + 20 mV and + 40 mV. Pipette resistance ranged from 4–5 MΩ and the internal solution consisted of the following: 145 mM CsMeSulfonate, 10 mM hydroxyethyl piperazineethanesulfonic acid (HEPES), 4 mM Na2ATP, 0.4 mM NaGTP, 5 mM QX-314, 4 mM MgCl2, 10 mM Na2phosphocreatine, 0.2 mM EGTA.4Na, 10 mM TEA, and 0.1 mM spermine dissolved in MilliQ water.
The stimulating electrode in stratum radiatum was placed 200–400 μm from the patch electrode in stratum pyramidale. Cells having resting membrane potentials of < 60 mV and exhibiting an input resistance of > 30 MΩ, and with an access resistance of 10–20 MΩ were included in the experiment. Cells were discarded if the access resistance changed by > 25% from their baseline values. Synaptic I/V plots were constructed by first, normalising each cell’s EPSC amplitude data as a percentage of the -60 mV response, and then plotting normalised average AMPAR synaptic current responses versus their respective holding potential from -70 to + 40 mV for each group. The Rectification Index (RI) was calculated from the synaptic I/V values as the ratio of EPSC amplitudes between -60 and + 40 mV. 1-Naphthylacetylspermine (NASPM) was used to selectively inhibit calcium-permeable AMPARs.
All EPSCs were recorded in voltage-clamp mode using a MultiClamp 700B (Molecular Devices) microelectrode amplifier and stimulation by the tungsten electrode was controlled through a custom-built programmable constant-current stimulator. Slices were imaged using differential interference contrast fitted to an Olympus BX50 upright microscope under a 40X objective. The following drugs were added to the aCSF: SR95531 (5 μM), gabazine (5 μM) and NASPM trihydrochloride (20 μM), all from Abcam; LY341495 (3 μM, cat# 1209), nimodipine (10 μM, cat# 0600), D-AP5 (100 μM, cat# 0106) and CGP55845 hydrochloride (1 μM, cat# 1248), all from Tocris.
Data analysispCLAMP version 10 (Molecular Devices) software was used for AMPAR-mediated EPSC data acquisition and analysis. Statistical significance was determined by univariate ANOVA followed by Bonferroni post hoc analysis for the different genotypes using Prism 9.0 software (Graph Pad Software). Data are reported as mean ± SEM.
ECoG measurementsPrior to surgery, mice were housed in groups of up to four animals. Mice that underwent ECoG electrode implantation surgery and recordings were housed individually during recovery and for the duration of recordings.
Electro-corticogram (ECoG) electrode implantation surgeryMice were surgically implanted at 40 weeks of age or older and allowed to recover for at least 7 days before recording. Isoflurane (IsoFlo; Abbott Laboratories) at a concentration of 4–5% mixed in O2 (vol/vol) was used for induction of anesthesia, maintenance was achieved with a 1–2% concentration. The depth of anesthesia was assessed by the absence of foot and tail pinch withdrawal reflex. A subcutaneous injection of meloxicam (1 mg/kg) dissolved in 0.9% saline was given prior to surgery. The mouse was placed in a stereotaxic frame (myNeuroLab, Leica Microsystems) and the scalp was shaved and sterilized with 80% ethanol. The skin was infiltrated with Lignocaine (1% ampules, Pfizer) and a 1 cm long incision was made. The skull was cleaned with 3% hydrogen peroxide solution (Pfizer) and four burr holes were drilled onto the skull. Epidural stainless-steel wire electrodes (Plastics One Inc., part no. MS 333-3A 0.005 inches) were implanted on the right frontal (reference electrode), left parietal (recording electrode) and occipital bone (ground electrode). An anchoring 1 mm screw was implanted in the occipital bone (extradural) for stability. The electrode pedestal and anchoring screw were embedded in methyl methacrylate dental cement (Catalog #1255710; Henry Schein Inc). Mice recovered in a Thermacage (Datesand Ltd) at 30 °C until fully awake.
Video-electrocorticographic recordingsRecordings were done via a tripolar cable (Plastics One Inc., Catalog# 335-340-3 0-SPR 80CM tripolar) and a commutator (Plastics One Inc., Catalog #8BSL3CXCOMMT) connected to a Grael EEG amplifier (Compumedics). The cables were connected to the head pedestal under light anesthesia (1–3% isoflurane for less than 5 min). Signals were band-pass filtered at 1 to 70 Hz and sampled at 256 Hz using Profusion data acquisition system (Compumedics) with simultaneous synchronized video monitoring (Profusion EEG4; Compumedics). Video recording was done using the Vivotek video server (VS8102) connected to an infrared day and night digital color camera (EVO2; Pacific Communications).
Mice were recorded for up to 5 days with the first 12 h of recording excluded. All ECoG traces were visually examined offline and events were manually counted. Spikes were defined as isolated events of an amplitude of equal or > 500 μV and a duration of up to 80 ms. The data was reported as the total number of spikes examined over a 24 h window. The presence of seizure activity was confirmed in video recordings.
KA-induced seizuresMice were 22 weeks of age (± 2 weeks) at the time of testing. Animals were administered kainic acid (KA; 25 mg/kg i.p.; Sigma-Aldrich Pty Ltd) and observed for 2 h. Mice were scored for seizure level based on the Racine scale ranging from 0–7 as per our previous method [58, 69].
Cobalt uptakeCobalt uptake experiments in mice (42 weeks of age ± 2 weeks) were conducted as previously described [58, 70].
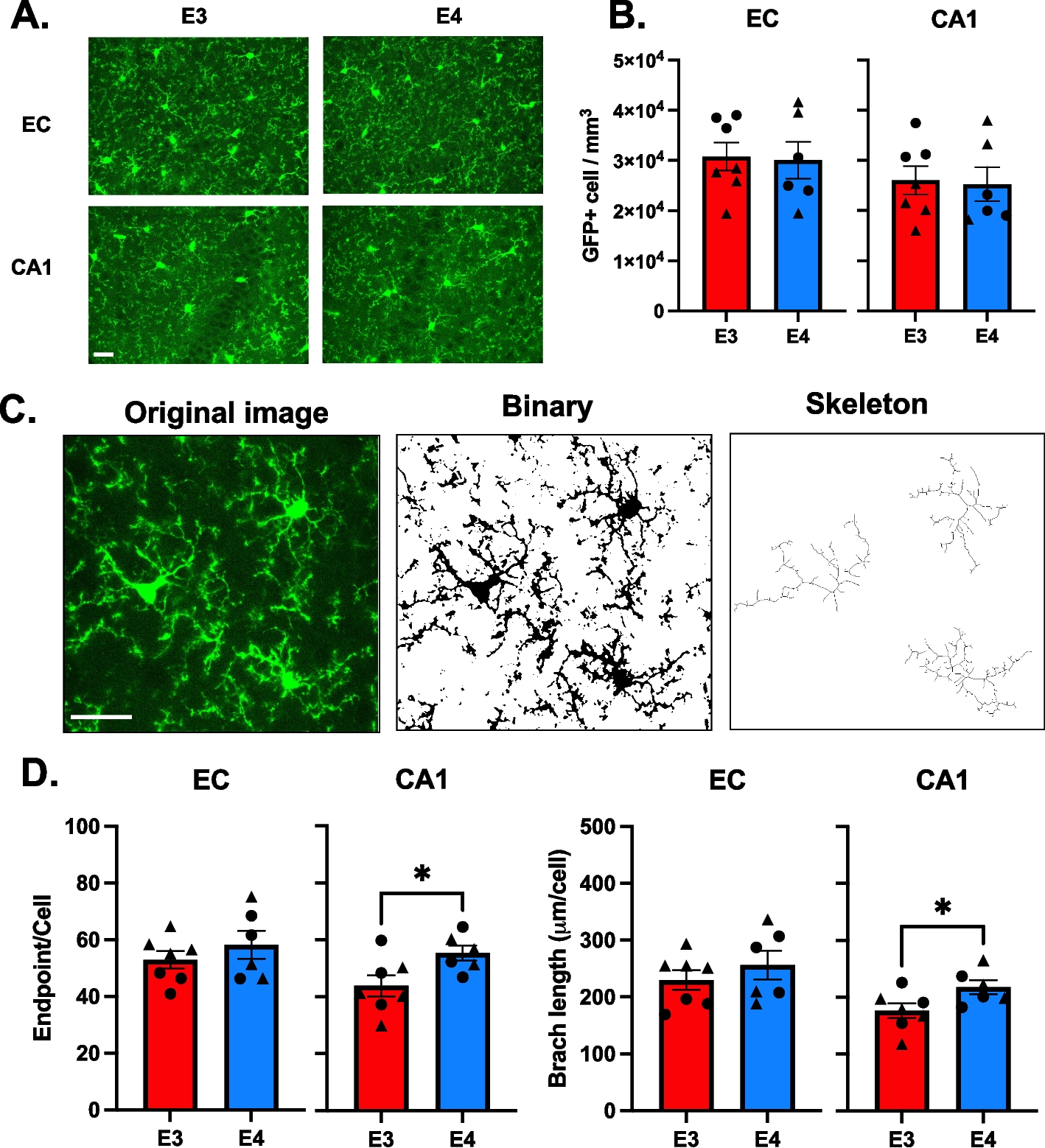
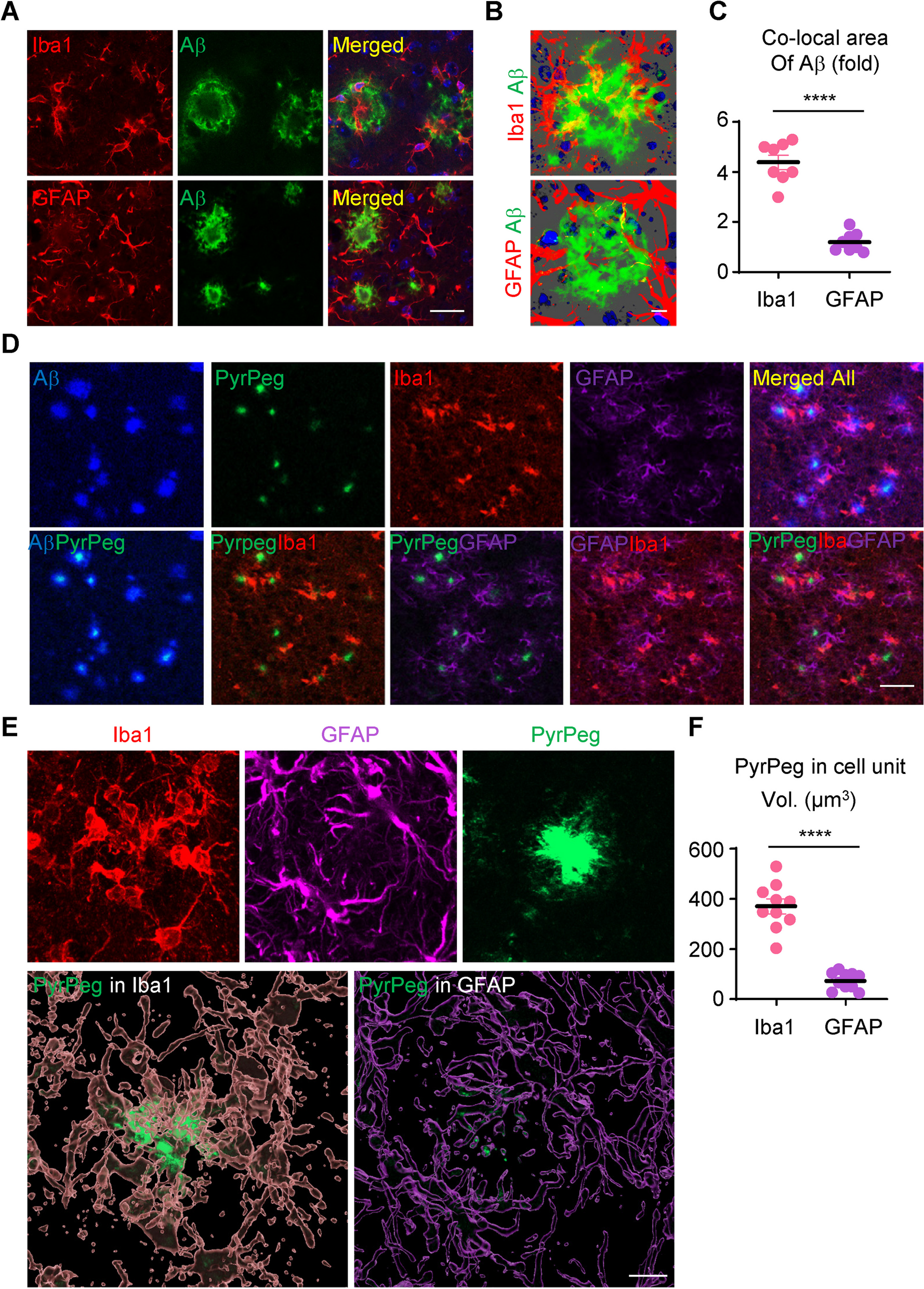
ImmunohistochemistryMice (42 weeks of age ± 2 weeks) were anesthetised with a cocktail of ketamine (8.7 mg/mL) and xylazine (2 mg/mL) via i.p. injection, and transcardially perfused with 4% PFA. Brains were coronally sliced at 40 μm thickness using a cryostat (Leica Microsystems CM3050 S) and immunohistochemistry on free-floating sections were conducted as previously described [58, 65]. Sections were incubated in the following primary antibodies: mouse anti-NeuN (1:500; Millipore Cat# MAB377, RRID:AB_2298772, Merck Millipore), rat anti-CD68 (1:100; Bio-Rad Cat# MCA1957, RRID:AB_322219, Bio-Rad Laboratories), rabbit anti-GFAP (1:300; Agilent Cat# GA524, RRID:AB_2811722, Agilent Dako) and human anti-6E10 (1:1000; Covance Cat# SIG-39345-200, RRID:AB_662802, BioLegend). Following primary antibody incubation, tissue sections were subsequently incubated in the appropriate biotinylated secondary antibodies (1:250; Thermofisher Scientific).
StereologyQuantification of cell population estimates were made using a brightfield microscope (Zeiss Axio Imager A1) as previously described [58, 65]. All stereological cell counts were performed blind to genotype and age.
Golgi impregnationUnder isoflurane gas anaesthesia, mice (42 weeks of age ± 2 weeks) were euthanized by cervical dislocation and brains were immediately removed. The brains were coronally sliced at 100 μm thickness using a cryostat (Leica Microsystems CM3050 S) and stained using the FD Rapid GolgiStain™ kit (FD NeuroTechnologies Inc, MD) as per the manufacturers’ recommendations. Sections were coverslipped with Permount™ (ThermoFisher Scientific) and allowed to dry for 24 h prior to analysis using a brightfield microscope (Zeiss Axio Imager A1). Dendritic morphology was assessed as previously described [58]. For each brain, 5 neurons from the hippocampal CA1 pyramidal layer were traced. CA1 spine density was assessed by counting the number of spines in 3 branches per neuron (5 neurons/brain) of branch orders 2–4 as previously described [58]. All protrusions < 2 µm were counted as spines provided they were continuous with the dendritic shaft. The spine density was defined as the number of spines per 10 µm of dendritic length.
Amyloid assessment6E10 quantificationQuantification of the 6E10 staining was performed using the Image-Pro Plus v.6.0 image analysis system to analyze the percent area occupied by positive staining, as previously described [65].
Amyloid-β plaque quantificationThioflavin S staining was used to determine the number of fibrillar Aβ plaques, as previously described [65]. WT mice were not assessed due to no detectable Thioflavin S being observed [71].
Amyloid-β ELISAsMice were cervically dislocated under isoflurane gas anaesthesia and the hippocampus was rapidly dissected, weighed and homogenised in 5 vol/wt of TBS (Tris-HCL 50 mM pH 7.6; NaCl 150 mM; EDTA 2 mM) containing a cocktail of protease inhibitors (1:100, Sigma-Aldrich Pty Ltd). Samples were then suspended in 2% SDS containing protease inhibitors (1:100, Sigma-Aldrich Pty Ltd) and centrifuged at 100,000 × g for 1 h at 4 °C. The supernatant containing the soluble Aβ fraction was collected. The remaining pellet was resuspended in 20 μl of 70% formic acid, homogenised and centrifuged at 100,000 × g for 1 h. Following centrifugation, 180 μL of Tris–HCL (1 M, pH 11) was added to neutralise the sample. The supernatant containing the insoluble Aβ fraction was collected. The Aβ levels were determined by using the BetaMark™ Total Beta-Amyloid Chemiluminescent ELISA Kit (Cat #: Covance Cat# SIG-38966-kit, RRID:AB_10718506; BioLegend), BetaMark™ Beta-Amyloid x-40 Chemiluminescent ELISA Kit (Cat #: SIG-38950; BioLegend) and BetaMark™ Beta-Amyloid x-42 Chemiluminescent ELISA Kits (Cat #: SIG-38952; BioLegend), as per manufacturers’ instructions. Previous studies have shown mice overexpressing hAPP, including J20 mice, exhibit a much larger signal in Aβ levels compared to WT mice [71, 72]. As such, only comparisons between J20 and Gria2G/G/J20 mice were conducted.
Cytokine ELISAsQuantification of the inflammatory cytokine TNF-α was conducted via antibody-specific ELISA. Mice were anesthetised with isoflurane, cervically dislocated, the hippocampus was removed, snap frozen and stored at -80 °C until use. The tissue was homogenised in 50 mM Tris-HCl, pH 7.2, 50 mM NaCl, 1% Triton X-100 and 50 mM Sodium Fluoride (NaF) containing protease inhibitors (1:1000, Sigma-Aldrich Pty Ltd). Samples were centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant was collected and total protein concentration was determined using the Bradford Assay. TNF-α (BioLegend Cat# 430901, RRID:AB_2883995) protein concentrations were quantified by ELISA kit in accordance with the manufacturer’s instructions.
Western blotsMice were anesthetised with isoflurane, cervically dislocated and the hippocampus was rapidly dissected and frozen at -80 °C until use. Unless otherwise specified, tissue was homogenised by sonication in 500 μL RIPA buffer (Sigma-Aldrich Pty Ltd) and supplemented with a protease inhibitor cocktail (1:100, Sigma-Aldrich Pty Ltd). Proteins were resolved by SDS-PAGE on 4–12% Bis–tris gels (NW04122BOX, Thermofisher Scientific) in 1 × MES SDS running buffer (B0002, Thermofisher Scientific), transferred to polyvinylidene difluoride (PVDF) membranes (IB24001, Thermofisher Scientific) and blocked with 5% non-fat dry milk for 1 h at room temperature. Membranes were immunoblotted with primary antibodies overnight at 4 °C with agitation followed by a 1 h incubation with the appropriate horseradish-peroxidase (HRP)-conjugated secondary antibody at room temperature with agitation. Signals were developed with chemiluminescence (WP20005, Thermofisher Scientific) and exposed to film. Where appropriate, antibodies were removed with stripping buffer (100 mM 2-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7) at 50 °C for 45 min, followed by washing for 1 h with tap water and re-probing membranes for β-tubulin. The intensity of bands was measured by using ImageJ software. The following antibodies were utilized: ADAR2 (1:1000, Santa Cruz Biotechnology Cat# sc-33180, RRID:AB_2222780), GluA1 (1:1000, Millipore Cat# AB1504, RRID:AB_2113602, Merck Millipore), GluA2 (1:1000, Abcam Cat# ab20673, RRID:AB_2232655, Abcam), GluA2/3 (1:1000, Millipore Cat# 07-598, RRID:AB_310741, Merck Millipore), GluA3 (1:1000, Cell Signaling Technology Cat# 3437, RRID:AB_1264115, Cell Signaling Technologies), β-tubulin (1:1000, Promega Cat# G7121, RRID:AB_430874, Promega), HRP-conjugated anti-rabbit IgG (1:5000, Millipore Cat# AP132P, RRID:AB_90264), and HRP-conjugated anti-mouse IgG (1:5000, Millipore Cat# AP124P, RRID:AB_90456).
Co-immunoprecipitationCo-immunoprecipitation experiments were conducted as previously described [58, 73]. Mice were anesthetised with isoflurane, cervically dislocated and the hippocampus was isolated and frozen at -80 °C until use.
BS3 crosslinkingBis(sulfosuccinymidal)suberate (BS3) is a cell-impermeable crosslinker which covalently bonds surface proteins to other nearby surface proteins thereby increasing their molecular weight. Specific crosslinked protein aggregates can then be identified via immunoblot detection. BS3 crosslinking therefore enables the discrimination of AMPARs located on the cell surface from intracellular AMPARs. BS3 crosslinking was performed as previously described [74]. Briefly, mice were anesthetized with isoflurane, cervically dislocated, the brain was rapidly removed, and the tissue was immediately mounted onto a vibratome. 400 μm coronal sections were taken from AP positions between bregma -1.34 mm and -2.3 mm. The hippocampus was then isolated from the slice, under a dissecting microscope. Hippocampal sections were added to 100 μL of ice-cold artificial cerebral spinal fluid (ACSF) that was immediately spiked with 2 mM of bis(sulfosuccinymidal)suberate (BS3; Thermofisher Scientific). For experiments conducted on CA1, CA3 and DG hippocampal regions, the regions were isolated under a dissecting microscope and incubated in ACSF with BS3 as above. Samples were incubated with agitation at 4 °C for 30 min, prior to the addition of 100 mM glycine for 10 min at 4 °C to terminate the reaction. Following crosslinking, tissue was centrifuged for 2 min at 17,000 × g to pellet the sample. The supernatant was discarded, and the pellet was resuspended in 200 μL of RIPA buffer (Sigma-Aldrich Pty Ltd) containing protease inhibitors (Sigma-Aldrich Pty Ltd) and homogenised by sonication. Samples were centrifuged at 17,000 × g. Samples were subjected to SDS PAGE and immunoblotting for GluA2 (Abcam Cat# ab20673, RRID:AB_2232655, Abcam).
Capillary electrophoresis and immunoprobingThese methods pertain only to supplemental Fig. 5b as this data was acquired post relocating to a new laboratory. Tissue was collected from 5xFAD WT and transgenic littermates at 40 weeks of age (± 2 weeks) by anesthetising mice with 5% isoflurane in air and transcardially perfusing with PBS. Brain tissue was hemisected and snap frozen before being dissected to isolate the hippocampus and suspending in 500 μl RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich Pty Ltd Cat#S8830) and PhosSTOP™ phosphatase inhibitors (Sigma-Aldrich Pty Ltd Cat#4906845001). Hippocampal tissue was then homogenised by low amplitude sonication for 20 s and incubated on ice for 45 min. Resulting cell suspension was centrifuged at 14,000 rpm for 15 min at 4 ℃ and the supernatant isolated and stored at -80 ℃. The optimal antibody concentration, and linear dynamic ranges for ADAR2 was determined prior to conducting expression analysis.
To quantify ADAR2 protein expression, samples were run on a standard 25-well WES operating plate, as per manufacturer’s instructions using the WES Simple Western instrument (ProteinSimple). Reagents were obtained from 12-230 kDa separation modules (ProteinSimple Cat#SM-W004) and Total protein detection modules (ProteinSimple Cat#DM-TP01). A protein concentration of 0.3 μg/μl was used for both wildtype and mutant hippocampal tissue. A working dilution of 1:30 was used for the ADAR2 antibody (Santa Cruz Cat#SC33180, RRID: AB_2222780, Santa Cruz Biotechnology) in all experiments performed.
For detection of ADAR2 expression, samples were replicated in two separate wells within individual plates and treated with either anti-ADAR2 primary antibody or the total protein assay. The total protein assay functions in a similar way to a Coomassie-stained gel whereby a biotin is attached to all proteins in the sample and incubation with streptavidin-HRP followed by luminol and peroxide generates a chemiluminescent signal for total captured protein. An internal control sample (derived from wildtype mice) was run in technical duplicates for both the primary antibody and total protein concentration, so that data could be standardised to this internal calibrator across different plates. Within each plate, several wells were used to control for variables including background biotinylation for sample diluent in the absence of sample, background biotinylation for the sample in the absence of the biotinylation label and background antibody signal for the sample diluent in the absence of sample.
Data was analysed using the WES instrument software (ProteinSimple, Compass for SW 4.1 Windows 7/8/10 64 bit). Peak analysis settings were performed on the electropherograms (EPG) as follows: Range: (1–250); Baseline: threshold (0.1), window (400), stiffness (0.1); Peak Find: threshold (10), width (9), area calculation (Dropped lines). Baseline adjustments were made to fit relative background chemiluminescence signals with all samples measured at identical conditions. Dropped line analysis was preferred over a gaussian fit model to adjust for interfering additional peaks and for better control of relative peak signal. The ADAR2 antibody peak was identified approximately between 92–98 kDa. The resulting signal from the total protein assay yielded a broad multi-peak EPG and the cumulative area under these peaks was measured as the expression of all protein in the sample against which the target protein was normalised.
Behavioural testingEach behavioural testing trial included animals from all four genotypes. Behavioural testing was conducted using three different paradigms (one test per day) with separate cohorts of mice beginning at 24 weeks of age. Paradigm 1 consisted of open field test (OFT) followed by object recognition, elevated plus maze (EPM), Y-maze and working memory radial arm maze (RAM). Paradigm 2 consisted of OFT, object recognition, EPM, Y-maze and rotarod. Paradigm 3 consisted of reference memory RAM. After completion of testing, animals were euthanized for tissue collection studies at pre-determined ages.
Open field testThe OFT was performed as previously described [58, 65]. Briefly, the arena (40 × 40 cm) was situated in a large sound-attenuating box and had clear plexiglass walls, no ceiling, and a white floor (Med Associates Inc). The total distance traveled over 10 min was recorded. The arena was thoroughly cleaned with 70% ethanol (EtOH) between each mouse.
Elevated plus mazeThe EPM was performed as previously described [65]. Briefly, the EPM consisted of four arms (77 × 10 cm) elevated (70 cm) above the floor (Med Associates Inc), two of which had 20 cm high walls (i.e. the ‘closed’ arms) and the remaining two had no walls (i.e. the ‘open’ arms). A video camera recorded the mouse and a computer software program (Limelight; Med Associates Inc) was used to measure the number of open arm entries and time spent in the open arms, as an indication of anxiety-like behavior. The ratio of open arm entries to total entries was analyzed.
RotarodMice were place on a suspended rotating beam (Med Associates Inc) and the total time spent on the beam was recorded over three trials (1 trial per day for 3 days), as previously described [58].
Object recognitionThe testing arena consisted of opaque plexiglass (50 × 30 cm) that was rectangular in shape, with 35 cm high walls. Two identical objects (red wooden blocks 6 × 4 × 3 cm in shape) were placed symmetrically 15 cm apart from each other and approximately 5 cm away from the arena walls. The protocol was similar to Heneka et al. [75], with slight variations; a single testing session, consisting of two trials 4 h apart, was conducted. During trial 1, the mouse was allowed to freely explore the identical objects for 10 min. During trial 2, the mouse was allowed to freely explore the arena for 5 min, however, this time, one object was replaced with the novel object (a wooden yellow arch 8 × 5 × 3 cm in shape). The arena and the objects were thoroughly cleaned with 70% EtOH between each mouse. All trials were video-recorded and the time spent exploring each object during each trial was recorded manually. Exploration was defined as directing the nose to the object at a distance of no more than 1 cm and/or touching the object with the nose. Data is presented as the discrimination ratio: time spent exploring the novel object/time exploring both objects.
Y-mazeThe Y-maze was performed as previously described [75], with modification. Testing was conducted in an opaque plexiglass Y-shaped maze consisting of three arms (40 × 4 × 17 cm) diverging at a 120° angle (Med Associates Inc). Each mouse was placed in the centre of the Y-maze and allowed to explore freely through the maze during a video-recorded 5 min session. The sequence and total number of arms entered was recorded manually. Arm entry was counted when the hind paws of the mouse had been completely placed in the arm. Percentage alternation was calculated as the number of triads containing entries into all three arms divided by the maximum possible alternations (the total number of arms entered minus 2) × 100. The maze was thoroughly cleaned between each mouse with 70% EtOH.
Radial arm mazeThe RAM consisted of eight arms (65 × 9 cm), extending radially from a central arena (35 cm diameter) and placed on a table elevated (90 cm) above the ground (Med Associates Inc). Each arm and the central arena were made of plexiglass, with enclosing walls made of clear plexiglass. Extra-maze cues consisted of the investigator, who was located in the same position for all trials, as well as large, fixed furniture around the room. The RAM was thoroughly cleaned with 70% EtOH between each mouse. Each food reward container was wiped with a small amount of sweetened condensed milk prior to the commencement of each trial to avoid the presence of olfactory cues. Additionally, the maze was rotated 45° after all mice had completed the trials for the day to avoid the use of intra-maze cues during training.
Working memory RAMMice were individually housed and restricted to 85% of their original body weight for 1 week prior to the commencement of RAM testing. On the first, second and third day, mice were habituated to the maze by being placed into the central arena, with one arm open and baited with sweetened condensed milk. Starting on the fourth day and continuing once a day for 12 days, mice underwent a working memory task, where all eight of the arms were baited with sweetened condensed milk. The training trial continued until all eight baits were retrieved or until 8 min had elapsed. Following testing, the mice were returned to their home cage. An investigator recorded the total number of entries into the arms and an error was marked when a mouse re-entered an already retrieved arm within the same trial. Data are presented as “Session”, consisting of 2 days (a total of two trials).
Reference memory RAMMice were individually housed and diet restricted to 90% of their original body weight for 1 week prior to the commencement of RAM testing. Reference memory RAM was performed twice daily for 24 days, as previously described [65]. An investigator recorded the number of successful entries into the baited arms (where the sweetened condensed milk was consumed) divided by the total number of entries made. Data are presented as “Session”, consisting of 2 days (a total of four trials).
Statistical analysisAll statistical analyses were performed using the statistical package Prism 9 (GraphPad). For normally distributed data, differences between means were assessed, as appropriate, by one- or two- way ANOVA with or without repeated measures, followed by Bonferroni post hoc analysis. For non-parametric data, Kruskal-Wallis ANOVA was used, followed by Wilcoxon matched pairs signed-rank test. To assess differences between two groups, a student t-test was used. For t-tests, data sets were first tested for normality, before using parametric or non-parametric tests. For parametric tests, an F test for variance was used to determine whether standard deviations were equal between groups. If unequal, Welch’s correction was applied, as indicated. For non-parametric tests, the Mann-Whitney test was conducted, as indicated. All data is presented as mean ± SEM for line graphs, or mean ± SD for all other graphs, as indicated. For all statistical tests, a p value of ≤ 0.05 was assumed to be significant.
留言 (0)