N2a cell culture and transfection
Mouse neuroblastoma cell line, Neuro-2a (N2a) was cultured and maintained at 37 °C, 95% relative humidity and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% non-essential amino acids (NEAA, Gibco), 1% L-glutamine, and 1% penicillin, streptomycin, and neomycin (PSN, Gibco). Cells were seeded in 24 wells plates on coverslips either for proximity ligation assay (PLA) immunofluorescence and DNA-FISH, or in 6 wells plates for biochemistry and ChIP/RT-qPCR. Cells were transfected with Lipofectamine LTX according to the manufacturer´s instructions (ThermoScientific). For NPM1 knockdown (NPM1 KD) and DOT1L overexpression (DOT1L OE) experiments, N2a cells were selected with 9.3 µg/ml Puromycin starting 24 h after transfection and for the remaining time until fixation or collection. Where indicated 10 μM EPZ5676 (Active Biochemicals) in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) was added to N2a cells for 72 h and refreshed every second day. DMSO was used as control treatment. The following plasmids were used for transfection: pLKO.1-shCTR (Sigma, Non Target #3), pLKO.1-shNPM1 (Sigma, TRCN0000115430), pLenti-CMV-HA-2A-GFP (Genscript), pLenti-CMV-DOT1L-FLAG-HA-2A-GFP (Genscript) and pVB-DOT1L-FLAG (Vector Builder).
Cell lysis, co-immunoprecipitation and immunoblots
For total cell lysate preparation frozen cell pellets were lysed in Lysis Buffer (10 mM Tris pH8, 1 mM EDTA, 1%SDS) supplemented with complete Protease Inhibitor cocktail (Roche-Diagnostics). Cells in Lysis Buffer were kept on ice for 30 min and chromatin disruption was done by sonication for 4 min in AFA 130 μl tubes (Covaris) using the Covaris E220. Cell debris was removed by centrifugation at 14.000 rpm 4 °C 5 min. The supernatant was collected and protein concentrations were quantified photometrically with Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad). 5–15 μg of cell lysates were loaded on 4–20% gradient SDS-PAGE (Bio-Rad). Proteins were transferred to PVDF membranes or to nitrocellulose membranes for Ponceau staining using the Trans-blot Turbo Transfer System (Bio-Rad) following the manufacturer’s instructions. Upon transfer, membranes were further processed for immunoblot following standard procedures.
For co-IP experiments, the cell pellet was resuspended in co-IP Buffer 1 (20 mM Tris pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.5% NP40) and cell lysate was left 30 min on ice. Debris was removed by centrifugation at 14.000 rpm 4 °C 5 min. The cell lysate was incubated with 2 μg of the indicated antibody bound to protein G dynabeads (1 μg antibody/10 μl beads, Invitrogen) overnight. The immunocomplexes were then washed with co-IP Buffer 1 three times and separated by SDS-PAGE. Immunoblotting was performed following standard procedures.
For endogenous NPM1/DOT1L co-IP, cell pellet (4*107 cells) was resuspended in co-IP Buffer 2 (20 mM Hepes pH8, 300 mM NaCl, 0.5% NP40, 10% glycerol, 1 mM EDTA) and the cell lysate was left 30 min on ice. Debris was removed by centrifugation at 14.000 rpm 4 °C 5 min. The cell lysate was equilibrated to 150 mM NaCl by adding drop by drop co-IP Buffer 2 without NaCl. 2 μg of the indicated antibody bound to protein G dynabeads (1 μg antibody/10 μl beads, Invitrogen) was added overnight and cell immunocomplexes were further treated as described before.
For NPM1 cross-linking with EGS (ethylene glycol bis(succinimidyl succinate), ThermoScientific) N2a cells were transfected using Lipofectamine 2000 and allowed to express DOT1L-HA-FLAG for 72 h. The cells were then lysed in co-IP Buffer 1 and incubated with either DMSO or 0.5 mM EGS for 30 min at 25 °C. Crosslinking was quenched with the addition of 0.025 mM Tris–HCl pH 7.5 for 15 min at 25 °C. DOT1L immunoprecipitation was performed using HA antibody as described above.
For nucleic acid digestion, the cell extract in co-IP Buffer 1 was treated with 20 μg of RNase (Promega) or 5U of DNase (Promega) for 30 min before performing the co-IP as described above.
Cell fractionation was performed by resuspending cell pellets in 500 μl Lysis Buffer (15 mM HEPES pH7.5, 10 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 250 mM Sucrose, 0.4% Igepal, 1 mM DTT, Protease Inhibitor cocktail) followed by incubation on ice for 20 min. Nuclei were centrifuged at 2.000 rpm for 10 min at 4 °C and the supernatant was collected as the cytoplasmic fraction. Nuclei were then resuspended in 100 μl Nuclei Lysis Buffer (10 mM HEPES pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, Protease Inhibitor cocktail) and incubated on ice for 5 min. Nuclei were pelleted at 15.000 rpm for 5 min at 4 °C and the supernatant was removed as the nucleoplasm fraction. The volume of the nucleoplasm fraction was adjusted to 250 μl with co-IP Buffer (20 mM Tris pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.5% NP40) with Protease Inhibitor cocktail. The chromatin pellet was then resuspended in 250 μl co-IP Buffer (20 mM Tris pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.5% NP40) with Protease Inhibitor cocktail and sonicated with the use of Bioruptor (Diagenode) with the following setting: cycle number: 9 + 9 Time ON: 30 s Time OFF: 45 s. The chromatin and nucleoplasm fraction were the used for co-IP as described before.
The following antibodies were used for co-IP or immunoblots: anti-DOT1L (1:1000, rabbit #77087 Cell Signaling), anti-DOT1L (1:1000, rabbit #90878 Cell Signaling), anti-HA-tag (1:1000, rabbit, #3724, Cell Signaling), anti-H3 (1:1000, goat, ab12079, Abcam), anti-NPM1 (1:1000, mouse, ab10530, Abcam), anti-GAPDH (1:3000, mouse, ab8245, Abcam), anti-Actin ( 1:5000, rabbit, ab179467, Abcam) anti-H3K79me2 (1:1000, rabbit, ab3594, Abcam), anti-H3K27me3 (1:1000, mouse, ab6002, Abcam), anti-H3K9me2 (1:1000, rabbit, ab1220, Abcam), anti-H3K9ac (1:1000, rabbit, ab4441, Abcam). Densitometric analyses were done with ImageJ software.
Immunofluorescence in cultured cells
Cells were seeded in multi-well plates and transfected as described before. Medium was removed and cells were washed 3 times with PBS. Fixation was performed with 4% paraformaldehyde (PFA, Life Technologies) in PBS for 15 min at RT. After PFA removal, cells were washed 3 times in PBS. Cells were permeabilized with 0.1% Triton-X100/PBS for 15 min and blocked in 10% horse serum/PBS 1 h. Primary antibody incubation was done overnight at 4 °C in blocking solution. Cells were washed 3 times in PBS and incubated with fluorescent secondary antibodies in blocking solution at room temperature for 2 h. After washing 3 times with PBS, cells were incubated for 5 min with DAPI (4′,6-diamidino-2-phenylindole, ThermoScientific) and washed 3 times in PBS. Coverslips were mounted on glass slides with fluorescent mounting medium (#S3023, DAKO). The following first and secondary antibodies were used: LAMINB1 (1:250, rabbit ab133741, Abcam), FIBRILLARIN-Alexa488 (1:250, rabbit, ab184817, Abcam) H3K79me2 (1:250, rabbit, ab3594, Abcam), NPM1 (1:1000, mouse, ab10530, Abcam), GFP (1:250, rabbit, ab290, Abcam), donkey-anti-goat-Alexa488 (1:500, A-11055, Life-Technologies) or -Cy3 (1:500, 705-165-147, Dianova), donkey-anti-rabbit-Alexa488 (1:500, 711-545-152, Dianova) or -Alexa594 (1:500, 711-585-152, Dianova), donkey-anti-chicken-Alexa488 (1:500, 703-545-155, Dianova), donkey-anti-mouse-Alexa488 (1:500, 715-545-151, Dianova) or -Alexa594 (1:500, 715-585-151), donkey-anti-rat-Alexa488 (1:500, 712-545-153, Dianova) or -Alexa594 (1:500, 712-585-153, Dianova) and donkey-anti-rat-AMCA (1:200, 712-155-153, Dianova).
Immuno-FISH
The immune-FISH cells were initially treated as for the immunofluorescence experiment with the following changes: PFA-fixed cells were permeabilized with 0.2% TritonX100 in PBS, 10 min, then wash with 1 × PBS, 3 times, 5 min; primary and secondary antibody were incubated in PBG 1 × blocking buffer (0,2% Fish Gelatin (#G7041, Sigma) and 0,5% BSA in PBS). After washing out the secondary antibody with 1 × PBG, twice, 5 min and with 1 × PBS, twice, 5 min, cells were fixed again with PFA 4% (Life Technologies) with triton 0.1%, 10 min RT, followed by incubation with glycine 10 mM in H2O, 30 min, RT. Cells were then washed with 1 × PBS, 3 times, 5 min. For each coverslip 20 μl of hybridization mixture (Formamide 70%, Tris HCl pH 7.4 10 mM, blocking reagent 1x (#11096176001, Roche)) containing 0.5 μM of each DNA probe for major satellite repeats (mSat 3a/3b IDT, Additional file 3: Table S2) were added on a glass slide and the coverslips transferred carefully on the drop without making bubbles. The slide was put directly on a metal thermo block at 80 °C, 5 min and then hybridized in a humidified chamber, 2 h, RT. The coverslips were then put back in a 12 wells plate. Washed with Wash solution I (Formamide 70%, Tris HCl 15 mM pH 7.4, BSA 0.15%), twice, 15 min; with Wash solution II (Tris HCl 0.1 M pH 7.4, NaCl 150 mM, Tween 20 0.1%), 3 times, 5 min and incubated with DAPI, 2 min, RT. After a brief wash with 1 × PBS coverslips were mounted with mounting medium (#S3023, DAKO). The following primary and secondary antibody were used: GFP (1:500, cicken, ab13970, Abcam), donkey-anti-chicken-Alexa488 (1:500, 703-545-155, Dianova).
Image acquisition, quantification and analysis
Images were taken in a Leica Sp8 confocal microscope with a 63X oil objective and a zoom factor of × 3 when indicated. The mean area and number of FBL (FIBRILLARIN) nucleolar stained structures and the mSat foci were quantified using ImageJ with the ‘Analyze particle’ option. Particle size was set at 0.2-3000µm2. A two-way ANOVA with Sidak’s post-hoc test was performed with GraphPad Prism Version 6.07 to assess for statistical significance.
Proximity ligation assay (PLA)
PLA was performed with the Duolink starter kit reagents (DUO92103, Sigma) according to manufacturer's instructions using antibodies against FLAG (goat, ab95045, Abcam) and NPM1 (mouse, ab10530, Abcam). After completion of the PLA protocol, we proceeded with incubation of a third antibody produced in rabbit to detect cellular compartments such as nucleolus with FBL (FIBRILLARIN, rabbit, ab184817 Abcam) or nuclear membrane with LMNB1 (LAMINB1, rabbit, ab133741, Abcam) following the procedure for immunofluorescence. Images were obtained with a Leica SP8 confocal microscope and processed with the LASX software.
H3K79me2 ChIP-seq and bioinformatics analysis
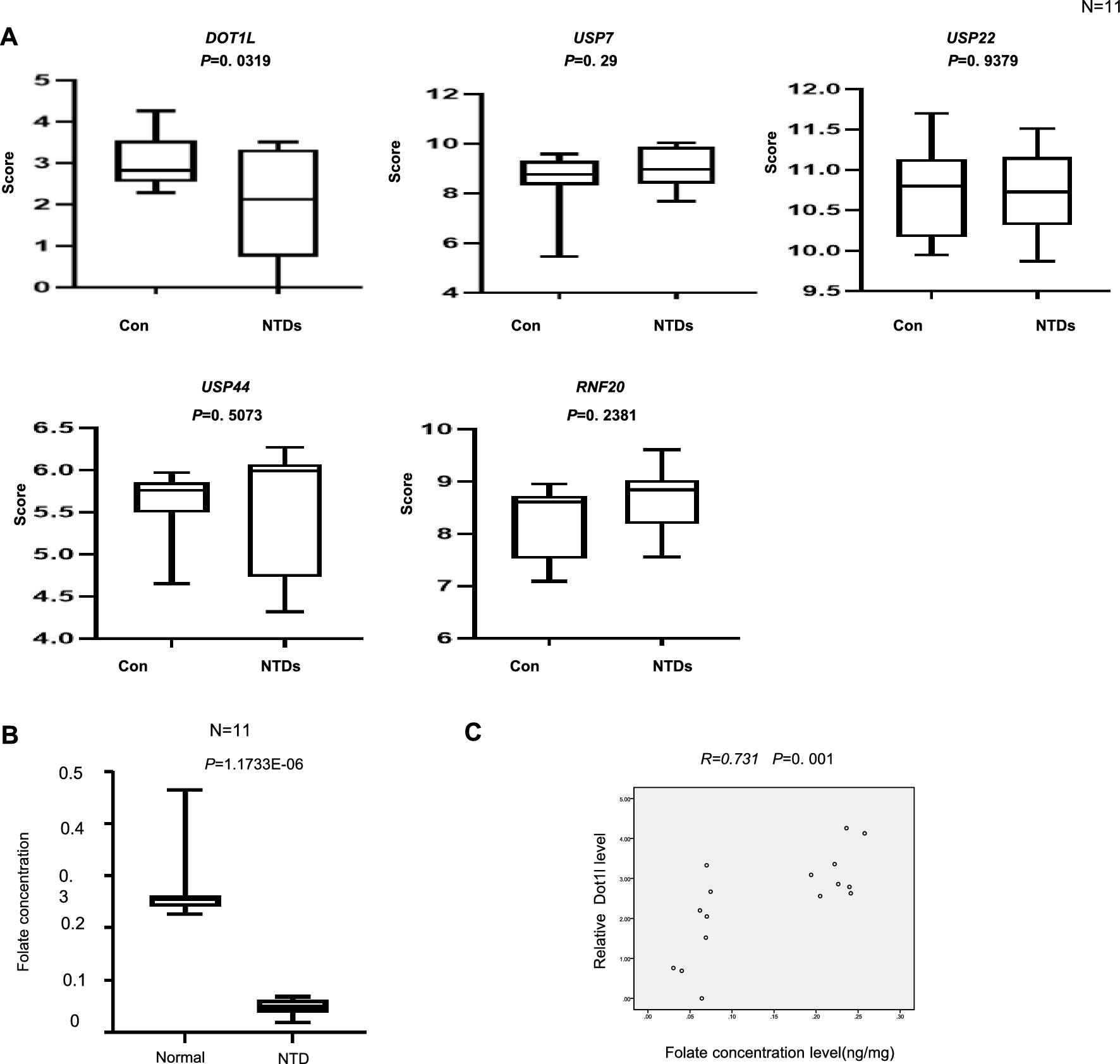
N2a cells were transfected with shNPM1 (NPM1 KD) or shControl plasmid (CTR) and were grown on 6 wells plates (~ 6 Mio cells) for 72 h. Two independent biological replicates per condition was used. Each biological replicate corresponds to a different passage of N2a cells and for each biological replicate transfection was performed in a separate batch. N2a cells were fixed with freshly prepared room temperature 1% PFA (Life Technologies) for 5 min at room temperature. Fixation was stopped with 125 mM glycine, and fixed cells were washed 2 times with ice-cold PBS. Cells were collected in ice-cold PBS and centrifuged 5 min at 500 rpm. Lysis Buffer (1% SDS, 10 mM EDTA, 50 mM Tris HCl pH 8.0, Protease Inhibitor cocktail) was added to the pellet and incubated on ice for 5 min. Shearing was done using Bioruptor with the settings: 3 × 10 min 30 s pulse, 30 s pause and chromatin was centrifuged for 5 min at 13.000 rpm and the chromatin samples were saved. Chromatin samples were precleared with protein G dynabeads (Invitrogen). Genomic DNA regions of interest were isolated using 4 μg of H3K79me2 antibody (rabbit, ab3594, Abcam). Complexes were washed, eluted from the beads with SDS Buffer (1% SDS, 0.1 M NaHCO3), and subjected to RNase and Proteinase K treatment. Crosslink was reversed by incubation overnight at 65 °C, and ChIP DNA was purified by Qiagen MinElute kit according to the manufacturer’s instructions. Quantification was performed by using PicoDrop and the PicoGreen quantification kit (Lumiprobe). Libraries were prepared using the NEBNext Ultra II DNA library preparation kit for Illumina (Biolabs) and sequenced using HiSeq 3000 (Illumina) (paired-end, 51 bp reads). Galaxy platform was used for quality control, mapping, peak calling and differential enrichment analyses [1]. Mapping of reads was performed on mouse genome build mm10 (GRCm38) using Bowtie2 (v2.3.4.1; [36]. High quality and uniquely mapping reads were retained (mapq > 5). MACS2 callpeak (Galaxy Version 2.1.1.20160309.6; [79]) was used for peak calling using default parameters. Only the common peaks in both replicates were retained to prevent false-positive peaks in downstream analysis. Diffbind [55] was used for differential peak enrichment analysis (NPM1 KD/Control) using the default parameters on Galaxy (Galaxy Version 2.10.0). The input of the analysis are peaks datasets and the aligned reads for each replicate in each condition, as well as the input controls. The package normalises, models the data, and performs differential binding analysis between the conditions specified (similar to DESeq2). Thus, all aligned regions are considered for the final calculation of fold changes and significance. Coverage was computed using multiBamSummary, and.bam files were normalized by bamcompare and bigwigcompare (deeptools, Galaxy Version 3.3.2.0.0; [53]. All metaprofiles and heatmaps of ChIP-seq signals were generated with deeptools (Galaxy Version 3.3.2.0.0. Peaks were annotated and visualized using ChIPSeeker (Galaxy Version 1.18.0; (T. [70, 71]. GO-term enrichment analysis was performed using clusterProfiler (R, v. 3.10.1; (T. [70, 71] (Yu, Wang and He, 2015)).
ChIP-qPCR
N2a cells were transfected with pVB-DOT1L-FLAG (Vector Builder) or pLKO.1-shCTR (Sigma, Non Target #3), pLKO.1-shNPM1 (Sigma, TRCN0000115430) as indicated and selected as described before. Chromatin was prepared as described for the H3K79me2 ChIP-seq, except for DOT1L ChIP by FLAG antibody. In this case, cells were treated with 1.5 mM EGS (ethylene glycol bis(succinimidyl succinate) for 30 min at RT before fixation with 1% PFA (Life Technologies) for 15 min at RT. The following antibody were used for the IP: H3K27me3 (rabbit, C15410195, diagenode); H3K79me2 (rabbit, ab3594, Abcam); FLAG (rabbit, 2368 s, cell signaling). Primers used for ChIP-qPCR are listed in Additional file 3: Table S2.
RT-qPCR
Total RNA was extracted using the RNeasy kit (Qiagen). Reverse transcription was performed using the RevertAid RT Reverse Transcription kit (ThermoScientific) following the user´s manual instructions. Primers used for RT-qPCR are listed in Additional file 3: Table S2.
Flow cytometry analysis of cell cycle
N2a cells (0.5–1 × 106 cells) were harvested after transfection and washed with PBS. Cells were resuspended in 0.5 ml of PBS and fixed by adding the cell suspensions drop by drop to 4.5 ml 70% ethanol and stored overnight at 4 °C. The fixed cells were centrifuged at 200 g 5 min RT and washed with PBS three times. Cells were then resuspended in 1 ml of freshly prepared PI/Triton X-100 staining solution with RNase A (0.1% Triton X-100, 0.2 mg/ml DNase-free RNase in PBS and 200 µl of 1 mg/ml PI (Molecular Probes)) and incubated for 3 h at RT before being analyzed by flow cytometry.






留言 (0)