
記住我





Human diploid cells need to handle a genome consisting of little more than 6 Gigabases in a way that protects it against damage and can be propagated unchanged over generations. Genes need to be activated and silenced depending on external signals, cell-fate decisions and to maintain cell identity. This depends on a tightly controlled access of gene-regulatory proteins to the genome. At the same time, the whole genome undergoes major reorganization in proliferating cells. The entire functional genome is duplicated during replication and then highly compacted upon entry into mitosis to allow segregation of sister chromatids and equal distribution of the genome onto the daughter cells.
The packing of the DNA with its bound proteins (chromatin fiber) within the cell nucleus is fundamental for these processes. In interphase, the chromatin of individual chromosomes locates in distinct chromosome territories with little intermingling. Genome-wide conformation capture studies such as Hi-C revealed that chromosomes can be divided into more transcriptionally active regions (A compartment) and inactive regions (B compartment). These are marked by specific histone modifications and interact with the same type of regions while avoiding the other compartment. The driving forces between this compartmentalization are complex and involve the folding of the chromatin fiber, general transcriptional activity and clustering of proteins and DNA, invoking mechanisms of phase separation. Liquid–liquid phase separation (LLPS) is a little understood multifactorial process underlying the self-organization of membraneless chromatin compartments, driven by intrinsic properties of the chromatin fiber, associated RNA and chromatin-binding proteins via disordered domains (e.g., BRD4) and/or by accumulation of transcription activators (transcription factories, RNA Pol II) (for a review see [1]).
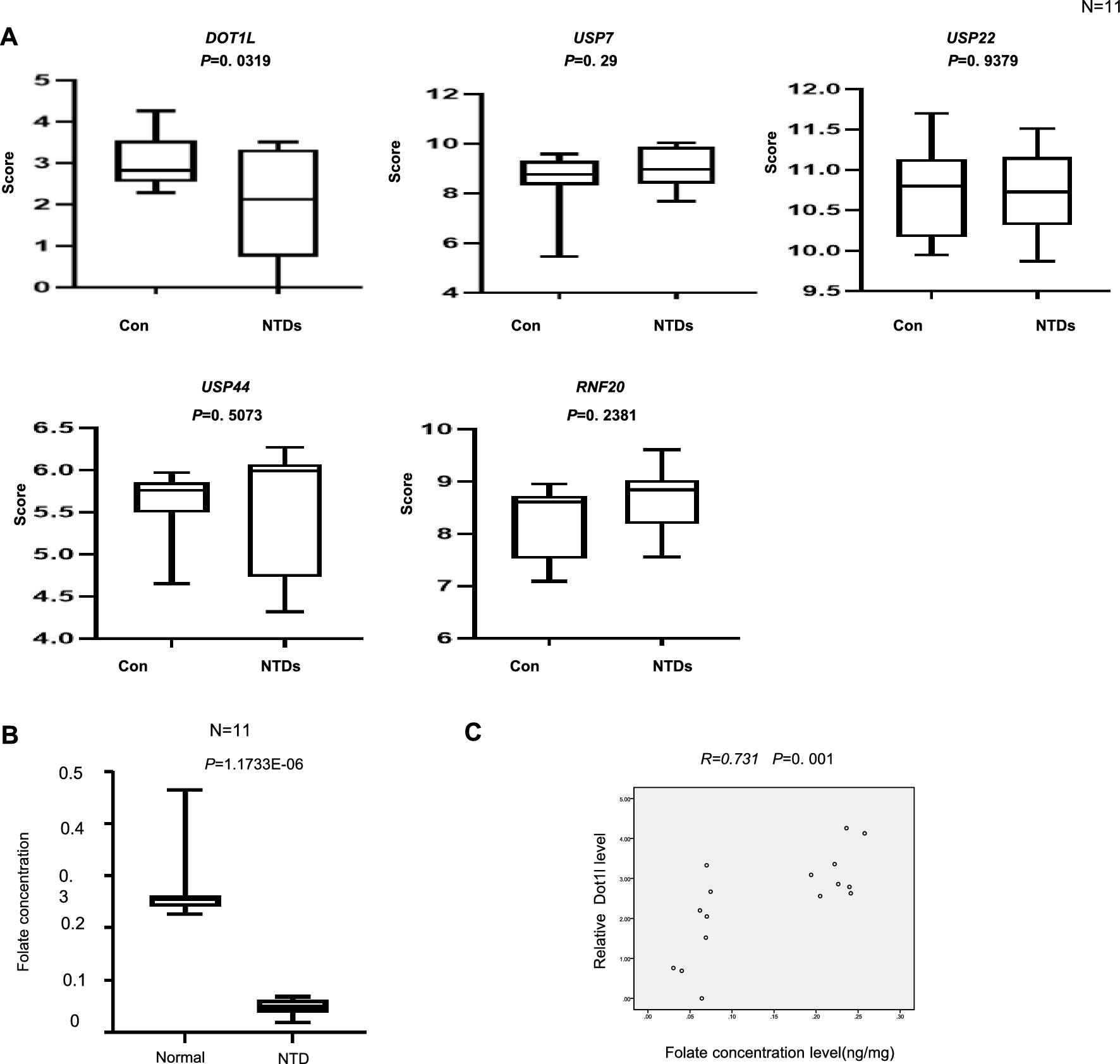
Chromatin conformation capture approaches revealed regions with strong directionality of chromatin contacts within these compartments, visible as characteristic triangles in contact maps and referred to as topologically associating domains (TADs) (Fig. 1). Contact maps obtained with higher resolution show that TADs consist of single or multiple contacts formed by distinct chromatin loops [2], which we refer to as “structural loops”, because they are very similar in different cell types. Methods like Hi-C generally process millions of cells and hence contact maps represent an average of a large cell population and do not yield information about the presence of the individual loops in individual cells.
Fig. 1
Organizational layers of chromatin and their reflection in Hi-C maps. Organizational layers of the chromatin fiber in the cell nucleus (top) aligned with the representation of those features in Hi-C interaction maps (bottom). Chromosomes reside in distinct territories with little intermingling. Therefore, chromatin contacts occur predominantly within the same chromosome (left panels). Within the chromosomes compartments with different chromatin properties (A compartment—active chromatin, B compartment—inactive chromatin) can be observed that interact with regions belonging to the same compartment type (blue box) (middle panels). Underlying these features in the folding of the chromatin fiber into chromatin loops, to a large extend mediated by the cohesin complex and CTCF, visible in the 1 Mb region shown for chr19 as regions with strong interactions (right panels). (Hi-C maps were generated using data for HCT116 cells by Rao et al. 2017 [5] and Juicebox.)
Key proteins for the formation of structural loops are the cohesin complex and the chromatin insulator protein CTCF. Depletion of critical cohesin complex subunits such as RAD21 or CTCF leads to the apparent loss of chromatin loops [3,4,5]. Models for the establishment of chromatin loops by cohesin and CTCF suggest an intriguing mechanism of ATP-driven loop extrusion by the cohesin complex that is halted when the extrusion complex encounters two convergently oriented CTCF sites [6, 7]. Within these structural loops more loops are formed between enhancers and promoters which we refer to as “functional loops”. Among the players involved in the formation and positioning of such loops are RNA polymerase II [8], the Mediator complex [9] transcription factors and cohesin. A reorganization of chromatin loops occurs during cell division. During chromatin compaction in the prophase and prometaphase of mitosis, the interphase loops are replaced by a more homogeneous type of loop compaction generated by condensin I and condensin II, loop-extruding complexes that are structurally and mechanistically similar to cohesin [10]. During entry into G1-phase the cohesin-dependent loops are re-established [11] with cohesin-mediated loop extrusion ongoing during the entire interphase. Thus, all types of chromatin loops are dynamic and are re-organized during the cell cycle and by transcriptional cues during differentiation and development.
The loops provide a dynamic structural scaffold to chromatin and impact gene expression, by directly recruiting enhancers to promoters or indirectly through confining the search space of enhancers.
A general depletion of cohesin or CTCF in cells only leads to mild misexpression of a few hundred genes [12], indicating that TAD chromatin loops are only one layer for controlling gene expression. However, disruption or reorganization of specific loops has been shown to impair development in mouse models [13] and to be linked to diseases, including cancer (e.g., enhancer hijacking in AML [14]) and to developmental defects [15].
Chromatin conformation capture methods (such as 4C, Hi-C, Micro-C or GAM) cannot show the dynamics of chromatin loops since they analyze millions of cells simultaneously; while available single cell Hi-C data [16] as well as super-resolution chromatin tracing with DNA-FISH [17, 18] indicate cell-to-cell variability with respect to presence and position of loops. Moreover, all these methods miss out the fourth dimension—time—since they are carried out on fixed cells.
The observation and eventual manipulation of chromatin contacts in live cells by imaging techniques provides a new perspective on chromatin dynamics. Fundamental questions can be approached from a different angle, for example the dynamics of promoter–enhancer interactions, the stability and duration of looping interactions, the role of proteins (in addition to CTCF and cohesin) and protein aggregates/droplets (e.g., transcription factories) for stabilizing or destabilizing specific loops and chromatin-reorganizing processes (replication and cell division). These observations require the labeling of DNA loci with fluorophores and live-cell imaging at high resolution. Major challenges include the selection of suitable labeling approaches to obtain a high signal-to-background ratio that permits visualization and tracking of the signals, to allow imaging over long periods of time (see also the review by the Hansen lab) [19]. In this review, we will discuss the different approaches successfully used to label DNA loci in live cells and highlight novel insights gained with these techniques.
Single locus labeling approachesFluorescent repressor operator systems (FROS)The first approach used to visualize specific sequences in mammalian cell nuclei was the lac operator–repressor system. In a pioneer study, Robinett et al. [20] inserted a mammalian DHFR expression vector with 256 lac repeats randomly into the genome of Chinese hamster ovary cells and demonstrated that the genetically amplified operator array gives a comparable chromatin staining after in situ hybridization, immunostaining with exogenous lac repressor protein after fixation and live cells by expressing the lac repressor protein fused to a fluorescent protein and a nuclear localization signal. Moreover, in the absence of gene amplification but with appropriate nuclear expression levels, a single copy insertion of 256 lac repeats provided sufficient signal-to-background signal to detect the locus as a diffraction limited spot (Table 1) [20].
Table 1 Summary of the advantages and disadvantages of the discussed DNA labeling methodsThe use of the Lac repressor–operator system was followed by the successful adaption of several other repressor–operator combinations (TetR, λcI, MalI and CymR) [21, 22] which allowed researchers to simultaneously visualize multiple genomic loci at diverse locations in live cells for the first time.
In these initial FROS experiments, repressor proteins were used because their high affinity to the operator binding sites would create a spot with a detectable signal-to-background ratio. However, it was shown later that the tight binding of LacR and TetR molecules to their operator sequences can have undesired biological consequences such as gene silencing. In the human osteosarcoma U2OS cell line, insertions of lac repeats were shown to induce an arrest in early S-phase, possibly by stalling DNA polymerase [23]. The same replication roadblock effect was induced by the introduction of a tet operator array in Escherichia coli [24]. In mouse embryonic stem cells, it was reported that the insertion of lac operator arrays next to the DHJH elements and the Eμ enhancer of the immunoglobin locus failed to result in germline transmission. The integration of the lac operator array in mouse embryonic stem cells resulted in an abnormal chromosome count of 70–80 chromosomes, indicating that the insertion of the lac operator array can lead to chromosome instability [25].
As shown by these examples, FROS labeling might induce genomic instability, and the effect of genomic insertion of FROS arrays needs to be carefully controlled.
Before the advent of CRISPR-based genome editing, the use of operator–repressor arrays to image specific loci of interest was largely limited to species with high rates of homologous recombination such as Saccharomyces cerevisiae [26]. In mammalian cells, FROS arrays were typically randomly inserted into the genome. With the increased possibilities of genome editing by homologous recombination and more recently CRISPR–Cas9 directed insertion of different operator–repressor assays within the same locus or chromosome became a possibility. However, the large size and repetitive nature of the operator arrays (typically 196–256 operator binding sites making up a total of 7–12 kb) still posed an experimental hurdle for facile CRISPR-based genome integration. Alexander et al. circumvented this problem by first integrating two AttP landing sites recognized by the bacteriophage integrases PhiC31 and Bbx1. This was followed by the integration of a plasmid containing the operator array sequence and an antibiotic resistance gene, which was removed later by CRE-lox or FLP-FRT recombination [27]. Tasan et al. developed a method of synthesizing a 96-mer TetO array with varying intermittent spacer sequences. This optimized TetO array of approximately 3 kb length can be inserted via CRISPR–Cas9 in a genomic location of interest and no perturbation of the nuclear localization or chromatin state of the targeted loci was observed. By employing a mutant TetR protein and mutated TetO sequence, even the multiplexing of two TetR type FROS systems in mammalian cells became possible [28].
Transcription activator-like effectors (TALEs)Transcription activator-like effectors (TALEs), discovered in the plant pathogenic bacteria Xanthomonas, are proteins that bind to specific DNA sequences [29]. TALEs bind to DNA through a domain of 32 to 35 amino acids called repeat-variable di-residues (RVDs) with two hypervariable amino acids, each of them binding to a specific base pair. Through DNA cloning, the amino acid code can be reprogrammed to bind specific DNA sequences. This versatility made TALEs a popular platform for applications such as gene editing and transcriptional modulation [30, 31].
Since TALEs can be modified to bind specific DNA sequences, it has also been an attractive method to fluorescently label such sequences (Table 1). Several studies have successfully labeled repetitive sequences like telomeres and centromeres in different circumstances. For example, Miyanari et al. demonstrated that TALEs can be used to study chromatin dynamics throughout the cell cycle [32]. Ren et al. have studied age-associated genomic alterations at telomeres and centromeres in premature aging models in both human and mouse. Others have labeled major satellite repeat regions and followed the dynamics of chromosomes through the cell cycle or were able to differentially label both parental chromosomes in hybrid mouse cells [31, 33].
While studies on repetitive sequences have provided valuable insights, it is still limited to a set of specific repetitive regions. To research TAD boundaries or enhancer–promoter interactions, it would be necessary to label non-repetitive single loci. Ma et al. demonstrated that TALEs can be used to visualize single DNA loci by mapping HIV-1 proviral DNA sequences in the human genome using TALEs labeled with quantum dots (QDs), which have unique optical properties including excellent brightness and photostability compared with traditional organic dyes [34]. The QDs greatly improve the signal-to-background ratio, reducing the number of fluorescent particles necessary to be detectable. However, a disadvantage is that TALE-QD particles need to be delivered to cells by transfection for each experiment, which limits their application.
While the other methods mentioned in this review are currently more prevalent, TALEs can be a valuable tool for DNA visualization. It might be interesting to develop a repetitive array, similar to FROS arrays, that can recruit a set of the same TALEs without the need to develop many different TALEs each targeting a specific DNA sequence. This would yield another system that can be used in combination with other DNA labeling systems such as FROS, dCas9 or ANCHOR. In addition, in cases were the binding of repressors to operator arrays induces replication roadblock effects, TetO targeting TALEs could be an alternative. An advantage of TALEs is that the residence time of TALEs can be adjusted by varying the length of the DNA binding domain, whereas repressors can currently only be altered by previously described mutations [35].
CRISPR/deadCas9 (dCas9)The CRISPR/Cas9 (Cas9 for short) genome editing tool that revolutionized biomedical research can be repurposed for labeling of genomic regions. Originally an antiviral defense system of prokaryotes, the Cas9 protein together with a guide RNA (sgRNA) sequence can induce a double stranded break with near base pair precision [36]. This made Cas9 the gold standard of genome editing [37]. Catalytic inactive mutants of Cas9 (deadCas9 or dCas9) turned out to be also quite versatile tools [38]. Since the dCas9 cannot cleave the target DNA, it is not released from chromatin. This long residence time makes it a very attractive tool to recruit proteins or protein domains, such as fluorescent tags or gene silencing domains, to specific genomic loci (Fig. 2). A clear advantage of dCas9-mediated labeling over other methods covered in this review (see Table 1) is that genome editing is in general not necessary, making it relatively straightforward to use on many different loci as well as in cell lines that are difficult to edit.
Fig. 2
Example image of ANCHOR and dCas9 labeling A: Image of a live HCT116 cell with two labeled loci, the D4Z4 repeats and a neighboring TAD boundary that are roughly 15 Mbp apart on chromosome X. The D4Z4 repeats were labeled with and a repeat-specific gRNA and dCas9-eGFP(green), the neighboring TAD boundary was visualized with an ANCH3 insertion in combination with the OR3-HaloTag protein and JF-549 dye (red). The image was taken on a SP5 Leica confocal microscope. B: Image of a live mouse embryonic stem cell with a locus on chromosome 4 visualized by an ANCH3 insertion and OR3-Halo/JF-646. The image was taken on a SpinSR SoRa Olympus spinning disk microscope. Note that the locus was already replicated and the two sister chromatids are visible
Chen et al. were the first to describe imaging of telomeres with dCas9-eGFP in combination with one sgRNA [39]. They also labeled the non-repetitive regions of the Muc4 gene with combinations of 26 or more sgRNAs. A similar approach was used to label enhancers and promoters with 36 sgRNAs each [40]. An alternative approach involving genome editing is the CRISPR-tag, developed by Chen and colleagues [41]. They inserted a repeat of short CRISPR target sites derived from the Caenorhabditis elegans genome (up to 6 repeats with at least 24 sgRNA sites) in the genomic region of interest [41] to recruit sufficient dCas9 molecules.
While it is very attractive to use dCas9 to label a specific locus, a major problem of dCas9-FP-based methods remains the low signal due to the limited number of fluorophores that can be recruited to the locus. Some groups succeeded in amplifying the signal by fusing the supernova tagging system (SunTag) to dCas9 [42,43,44]. The SunTag is a peptide scaffold with multiple GCN4 epitopes (e.g., 24) that can be bound by a single-chain variable fragment of an anti-GCN4 antibody. The nanobody can be tagged with fluorophores and expressed in mammalian cells (scFv-GCN4-GFP). When the SunTag system is fused to dCas9, this enables a substantial amplification of the fluorescent signal. This technique has been shown to work with guideRNAs for repetitive sequences as well as on non-repetitive regions such as the MUC4 gene [39, 45,46,47].
Alternative approaches that allow the recruitment of a large number of fluorophores per dCas9 molecule involve extending the sgRNA with RNA aptamer repeats such as repeats of the MS2 RNA binding sequence, bound by fluorophore-fusions of the MS2 bacteriophage coat protein, or other analogues [48,49,50,51]. Each RNA aptamer recruits two fluorescent proteins, leading to an accumulation of fluorescent signal at the target sequence. Combining different RNA aptamer analogues enables multi-color imaging. Maass et al. successfully labeled two different alleles in 129S1/CAST hybrid mouse embryonic stem cells (mESCs) and mouse embryonic fibroblasts (MEFs) by using single nucleotide polymorphisms in PAM sites [52]. This allowed them to confirm that single loci as well as chromosomes are stably positioned in space and time. Kamiyama et al. developed a splitGFP-system consisting of strands 1–10 of the GFP beta-barrel structure that only becomes fluorescent when it is complemented by the 11th beta-strand, which can be fused as a tagging sequence to a protein of interest [45]. Ghosh et al. [46] developed the GFP enhancer nanobody array (ArrayG) that can be attached to dCas9. Wild-type monomeric GFP molecules are initially dim but increase about 26 times in brightness when bound to the nanobody array. This system was used to track a repetitive genomic locus in live cells [47]. Clow et al. successfully adapted a dCas9-based platform that they previously used to label repetitive sequences (Casilio) [50, 53]. This system uses PumilioFBF (PUF)-tethered factors which can be engineered to bind to a specific 8-mer at the 3’end of a gRNA. By multiplexing the 8-mer sequences, PUF domains fused with fluorescent domains can accumulate at the dCas9. With this approach, they successfully labeled three different non-repetitive loci with three different colors. They demonstrate the applicability of Casilio for chromatin interaction studies by labeling the IER5L promoter and its super-enhancer, a known ~ 500-kb chromatin interaction dependent on RAD21 [5]. After degradation of RAD21, they indeed detected an increased distance between the labeled loci. The adaptations mentioned above use fluorophores (antibody-FP, MCP-FP and PUF-FP) which stochastically bind to the dCas9 fusion protein. This renders these systems inherently more resistant to photobleaching than dCas9-FP labeling schemes, paving the way for acquiring longer time tracks.
The advances on the dCas9 labeling techniques have significantly improved its signal-to-background ratio, resistance to photobleaching and ability to visualize loci over long time periods. Successful dCas9 labeling in these systems requires the expression of multiple genetic components (dCas9, binder and sgRNA(s)) either via transient transfection or the generation of stable cell lines. While dCas9 imaging systems can be relatively easily established on telomeres or other repetitive genomic loci, the step to label a single, non-repetitive locus is often large. This can be explained by the delicate balance of getting both a sufficient fluorescent protein level to label the locus and avoiding a high background signal that could obstruct the single locus signal. Expressing the fluorescent moiety at a low but sufficient level is therefore almost always an advantage in obtaining a high signal-to-background ratio, because expressing the fluorescent molecule at high levels increases the number of molecules in the background while the number of locus-binding molecules remains the same (small) number, certainly when single non-repetitive loci are targeted. At best the binding equilibrium at the target is somewhat improved, but this will be negligible relative to the increase in background fluorescent molecules, particularly when the fluorescent molecule has a long residence time at the target sequence. The flexibility of the dCas9 system has a great appeal for single locus tracking techniques, and ultimately a combination of solutions as described above might improve robust labeling of single loci.
The ANCHOR DNA labeling systemIn 2014, Saad et al. introduced a novel protein–DNA imaging approach called the ANCHOR DNA labeling system [54]. This labeling approach leverages the ParB–ParS binding of the ParABS partitioning system naturally occurring in bacteria. In the ANCHOR DNA labeling approach, fluorescently labeled ParB molecules (ORs) recognize a small set of ParS sites within a short non-repetitive INT sequence (ANCH) which can be integrated next to the locus of interest. Upon recognition of the ParS site by a ParB homodimer, a conformational change of the ParB molecules leads to N-terminal mediated oligomerization and spreading of ParB along the adjacent DNA strand via non-specific interactions. Together, the binding and spreading of fluorescent ParB proteins creates a diffraction limited spot at the locus of interest (Fig. 2) [55, 56]. A key advantage of the ANCHOR DNA labeling system is that it has been reported to not inhibit transcription even when present directly adjacent to a promoter or an intronic enhancer [55, 57].
Since different bacteria strains and replicons (e.g., genomes and plasmids) within the same bacterium contain different ParB-INT combinations, multiplexing non-crossreactive ANCHORs to label multiple loci became possible. For example, the ANCHOR1 and ANCHOR2 system were used in combination to visualize the dynamics of double strand break repair in Saccharomyces cerevisiae. Later, the ANCHOR1–ANCHOR3 pair was used in mouse embryonic stem cells to investigate the heterogeneity of chromatin dynamics [58]. Recently, significant differences between the labeling efficiency of different ANCHORs have been reported. A survey of single locus tracking techniques in Drosophila showed that ANCHOR2 labeled two regions of interest (ROIs) equally efficiently as LacI-LacO labeling, whereas ANCHOR1 showed less efficient labeling at both ROIs. In addition, both ANCHOR systems showed sensitivity to the design of the ParB-FP construct and cellular context [57]. This suggests that the extent of binding and/or cis-spreading of different ANCHORs might be tissue-, cell- and locus-dependent.
The ANCHOR DNA labeling system shows great promise as the latest addition to the available single locus tracking techniques. The ease of CRISPR-based genomic insertion of the short ANCH sequences (~ 1 kb) in combination with the ability to non-invasively label genomic regions of interest make the ANCHOR DNA labeling system a promising tool for studying locus dynamics (see Table 1). In the future, optimizations to the ParB-FP fusion design might further improve the labeling efficiency and robustness of the existing ANCHOR systems and novel ANCHOR types and combinations may be developed.
留言 (0)