
Remember me


The story began more than 180 years ago when Hünefeld detected crystals in squashed blood [1]. Probably, the crystals had been formed by hemoglobin, the ubiquitous red dye coloring blood. The identity of these crystals was confirmed 22 years later by Hoppe-Seyler [2], who isolated and crystallized hemoglobin. Today, such crystals would certify the uniformity of all hemoglobin molecules. At that time, however, things were less clear. In 1869 the nucleic acids were detected by Miescher [3]. In contrast to proteins, the nucleic acids did not crystallize. Analyzable crystals of these molecules were produced only almost 100 years later (Fig. 1).
Fig. 1
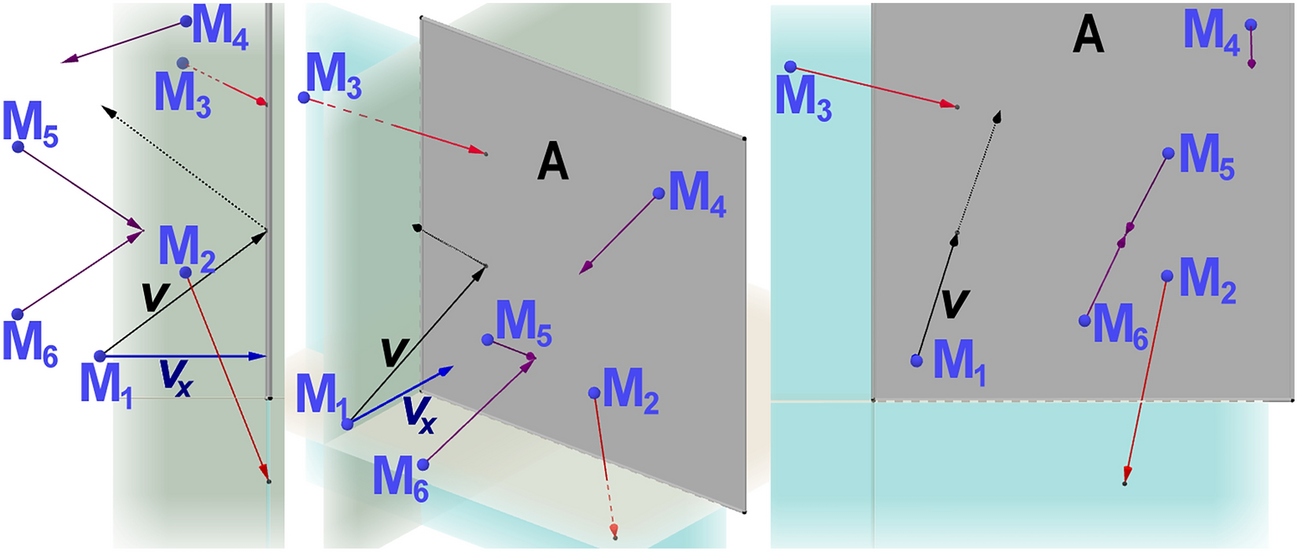
Six lines showing the method developments for structural analyses: red, direct methods for crystals of molecules below about 1500 Da; black, MIR and molecular replacement for protein crystals; green, MIR for crystals of nucleic acids; violet, reconstruction from electron microscope data; orange, calculation from nuclear magnetic resonance distances; blue, calculations based on the available sequences and structures in data banks. Asterisks indicate Nobel prizes. MIR multiple isomorphous replacement, CASP critical assessment of (protein) structure predictions (15 meetings to date)
In 1895 Röntgen [4] performed experiments with cathode ray tubes, in which electrons with energies of more than 10,000 eV were shot onto a metal like tungsten, where they produced a penetrating (so-called) X-radiation. This radiation showed, e.g., the bones of a hand on a scintillation screen without hurting the tissue. Consequently, this observation was developed into an important diagnostic medical tool. When in 1912, a crystal was exposed to a thin X-ray beam, a multitude of weak beams split away from the incident beam giving rise to so-called reflections that were documented on a photographic film. This observation was correctly interpreted by von Laue [5] as an interference phenomenon involving the scattering of an electromagnetic wave by the periodically located electrons in the crystal. This confirmed both the wave nature of the X-rays and the periodic arrangement of atoms in the crystal.
The wavelength of the X-rays was around 10–10 m = 1 Å, which corresponds to the atom–atom binding lengths and should therefore allow one to locate individual atoms in a crystal. The reflections are measurable because the scattered waves of millions of crystal unit cells add up in the interference process. Moreover, von Laue [5] showed that the electron density in the crystal (and thus the atom positions) can be calculated from a Fourier synthesis of the reflections. However, such a reconstruction requires the intensities as well as the phases of the reflections. Unfortunately, the phases cannot be measured directly, but only derived indirectly.
The phases may be established by combining several pieces of information: for instance, the electron density distribution in the crystal has to be positive everywhere, the distribution of electrons around an atomic nucleus is radial, the atom radii are known, all atomic bonds are close to a certain specific distance, the internal symmetries of crystals cause restrictions to the phase angles, partial structures may be known and accounted for (e.g., a phenyl ring), etc. In 1986, all these possibilities were compiled by Hauptman [6] under the name direct methods. Moreover, for crystals with less than a handful of atoms in the unit cell, a Fourier synthesis of the mere intensities without the phases (a Patterson function) may yield the positions of these atoms in the unit cell.
The first crystal structures, namely those of NaCl and diamond, were determined in 1913 by Bragg and Bragg [7]. They were followed by numerous other structures of larger molecules culminating in the structure of vitamin B12 (Mr = 1355), which was elucidated in 1956 by Crowfoot-Hodgkin [8]. Crystals of molecules smaller than vitamin B12 are usually analyzed by direct methods [6], but they do not work for larger molecules.
Around 1920, the intrinsic stability of proteins like hemoglobin was generally accepted, but proteineous enzymes remained mysterious. As enzymes catalyze chemical reactions, they should be intrinsically mobile. At that time, they were considered colloids without a stable spatial structure. The puzzle was solved in 1926 when Sumner reported crystals of the enzyme urease [9]. The crystals indicated that enzymes also have a defined spatial structure. Later on, it became clear that enzymes are indeed mobile, but can crystallize in one of their stable states. In 1995, the first movie of all states of an enzyme over a complete catalytic cycle was published [10].
The first structural knowledge on proteins did not come from crystals but from peptide fibers. In 1931, Astbury analyzed such fibers and detected two dominant X-ray scattering patterns, which he named α (observed with wool) and β (characteristic for silk) [11]. The actual structures of the α- and β-fibers remained obscure for 20 years. However, when Pauling studied the crystal structures of small peptides, he recognized that the bonds between the amino acid residues are always in the trans conformation, greatly restricting the structures of longer peptides [12]. Long all-trans-peptides can assume only two regular conformations stabilized by hydrogen bonding, the α-helix and the β-sheet, which actually corresponded to the α- and β-patterns of the fibers analyzed by Astbury [11]. These regular conformations turned out to constitute the dominant substructures (so-called secondary structures) of proteins.
The first serious X-ray diffraction experiment on a protein crystal (Fig. 2) was performed in 1934 by Bernal [13]. The crystals contained the enzyme pepsin and showed defined reflections up to high (about 2 Å) resolution, confirming the proposal of Sumner [9] and indicating that the atomic structure of pepsin could be obtained in principle. Actually, however, the protein structure remained unknown because the phases of the reflections could not be determined. A suitable method for phase determination was invented only 17 years later by Bijvoet [14], who compared the reflection intensities of the isomorphous crystals of strychnine sulfate and strychnine selenate and derived the position of the sulfur (selenium) atom in the unit cell by a Patterson function of the reflection intensity differences. The position helped decisively in determining all phases. Bijvoet named this the method of isomorphous replacement.
Fig. 2
A photograph of the first analyzed protein crystals as provided by the Svedberg laboratory in Uppsala/Sweden. The crystals consist of the enzyme pepsin and are up to 2 mm long. The photo is from the Bernal laboratory in London, courtesy of Judith A. Howard (Durham, UK)
Three years later, Perutz [15] used a variation of this idea with hemoglobin crystals. He soaked the crystal with a solution of mercury ions that bound locally in a defined manner at the free cysteines of the protein. Soaking was possible because his protein crystal, like all others, consisted of about 50% water. As usually several cysteines were available, he called this method multiple isomorphous replacement (MIR). The localized 80 electrons of a mercury atom change all reflection intensities measurably. A Fourier synthesis of these differences (difference Patterson) reveals the mercury atom positions, which in turn can be used for determining all phases. The MIR method was applied in almost all following structure analyses of proteins and nucleic acids. Astonishingly, Perutz [15] did not quote Bijvoet [14], the initiator of this method.
Using the MIR method, Kendrew [16] produced the electron density map of a myoglobin crystal (Mr = 17,000) 6 years later. During this analysis the phases of around 10,000 reflections had been calculated, which was an extraordinary logistic achievement in those days without versatile computers. It should be noted that the reliable interpretation of the resulting electron density map required the amino acid sequence of myoglobin. After the pioneering work of Sanger [17], that sequence was available on time. It turned out that myoglobin consists exclusively of α-helices, the geometry of which confirmed the substructure proposal of Pauling [12]. Five years after the atomic structure of myoglobin, Phillips [18] determined the first structure of an enzyme, lysozyme, which had crystallized in one of its stable conformations as proposed by Sumner [9] and Bernal [13]. In the beginning, the MIR phasing method was generally applied. However, after numerous protein structures were established, the molecular replacement method, in which phases were determined in a refinement using a resembling (part of the) protein structure, became popular [19].
The 60 years following the determination of the structure of myoglobin saw a multitude of reports on atomic protein and enzyme structures, giving rise to a very large amount of structural data. Since 1971, the protein structure data were normalized and compiled in an easily accessible bank, the Protein Data Bank [20, 21]. This bank brought an exceptional stimulus for this field of research.
Until 1985, all structures were from soluble proteins, because membrane proteins failed to crystallize as they associated nonspecifically at hydrophobic surface patches. After tedious experiments, Michel [22] observed in 1982 that membrane proteins can also be crystallized if their hydrophobic surface regions were covered by detergent molecules. This expanded the field of known atomic protein structures appreciably.
The size and the importance of the published protein structures grew with time. A typical structure is shown as a ribbon plot in Fig. 3. It is the membrane channel MspA, which is the base of the modern DNA sequence analysis [23, 24]. Several important atomic structures were rewarded with a Nobel prize, beginning with the first membrane protein [25], followed by the F1-ATPase [26], the potassium channel [27], RNA polymerase [28], and the G-protein-coupled receptor [29]. The analysis of crystallized proteins remains important because only this method allows for positional accuracies of 0.1 Å that are required for the explanation of catalytic processes.
Fig. 3
Ribbon model of the octameric membrane pore MspA from Mycobacterium smegmatis. The pore was produced efficiently by overexpression into Escherichia coli inclusion bodies and subsequent naturation [23]. As its pore diameter allows for the passage of a single-stranded DNA molecule, MspA was used for converting DNA sequence analysis into an automatic and cheap method [24]
Comments (0)