All unlabeled amino acids used in this study were of analytical grade and purchased from Carl Roth (Germany) or Sigma-Aldrich (USA). All single 15N labeled amino acids as well as labeled α-ketoisovaleric acid (3-methyl-13C, 3, 4, 4, 4-D4) (KIV) were purchased from Cambridge Isotope Laboratories, Inc. (USA). Labeled 4-methyl-2-oxovaleric acid (4-methyl-13C, 3, 3, 4, 5, 5, 5-D6) (MOV) was synthesized as reported (Lichtenecker et al. 2015). 6-diazo-5-oxo-L-norleucine (DON), aminooxyacetate (AOA), and D-malate (DM) were obtained from Sigma-Aldrich (USA).
Genetic engineering of E. coli A19
Gene deletions in A19 were performed according to Datsenko and Wanner (Datsenko and Wanner 2000) as adapted by the protocol of the Gene Bridges Quick and Easy™ gene deletion kit. FRT-PGK-gb2-neo-FRT cassettes with flanking regions homologous to the target genes were generated by PCR and generated kanamycin resistance (KanR) if integrated into the A19 chromosome. First, A19 cells were transformed with the pRed/ET plasmid and cultivated at 30 °C to an OD of approx. 0.6 in reaction tubes with punctured lids and shaking at 700 rpm. Expression of λ phage recombination proteins was induced by addition of arabinose to a final concentration of 0.3% (w/v) and incubated at 37 °C for 45 min. Subsequently, cells were cooled to 4 °C, harvested by centrifugation at 10,000×g, 4 °C for 1 min, washed three times with 1.5 mL ice-cold 10% (v/v) glycerol and harvested by centrifugation at 10,000×g, 4 °C for 1 min. The cell pellet was suspended in 40 µL 10% (v/v) glycerol solution and 1–2 µg linear FRT-PGK-gb2-neo-FRT cassette flanked by specific homology regions was added to the suspension before electroporation at 1450 V and ~ 5 ms pulse duration. Primers used for cassette generation are listed in Table 1. Subsequently, LB medium was added to the cells which were then incubated at 37 °C for another 3 h before being plated on kanamycin agar plates and incubated at 37 °C for 16–48 h. pRed/ET plasmid was cured during 37 °C incubation steps. Mutants were screened via colony PCR using KanR and gene locus specific primer pairs or primers annealing up-and downstream of the target gene (Table 1). Additionally, PCR products were purified and sequenced. For marker recovery, clones were transformed with 708-FLPe plasmid carrying FLP recombinase and plated on agar plates containing kanamycin and chloramphenicol. Clones were picked and grown in LB medium at 30 °C, to an OD of 0.6. A temperature shift to 37 °C induced the expression of FLP recombinase, which removed the marker from the gene locus via FRT recognition sites. FLP plasmid was cured during cultivation at 37 °C. Recovery was confirmed by PCR of the gene locus by subsequent PCR product purification and sequencing. Growth media were supplied with L-Gln or glucosamine if required for specific mutants. The "Stablabel" mutant containing mutations of asnA, ansA, ansB, glnA-his, aspC and ilvE was deposited in the German collection of microorganisms and cell cultures under the accession number DSM 116065.
Table 1 Primers used for E. coli A19 mutagenesis. “KO”-primers are used to generate a KanR cassette flanked by regions for homologous recombinationPreparation of S30 CF lysates
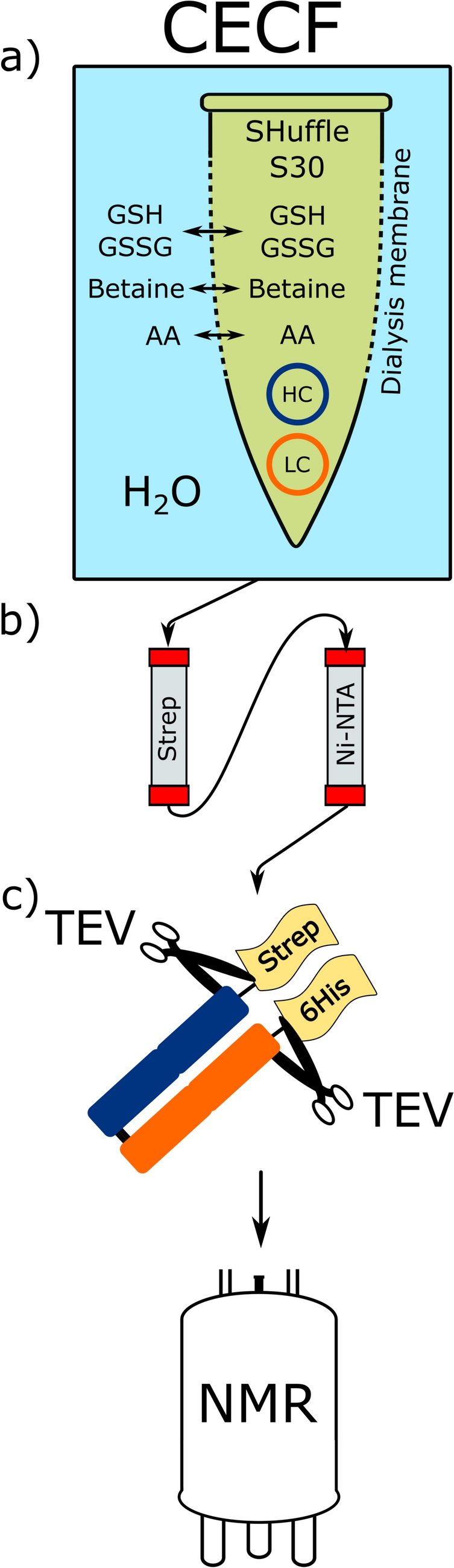
To produce lysates for CF expression, E. coli A19 cells were grown in 10 L YPTG (10 g/L yeast extract, 16 g/L tryptone, 5 g/L NaCl, 0.1 M glucose, 4.4 mM KH2PO4, 8 mM K2HPO4) tempered to 37 °C, 500 rpm and high aeration corresponding to ~ 3 reactor volumes of air per minute. Cells were grown until the culture reached an OD600 of 3.6–4.2 and then cooled down to 19 °C within 20 min. Subsequently, cells were harvested at 4500×g, 4 °C for 20 min and washed three times by centrifugation and suspension in 300 mL S30 A buffer (10 mM Tris–HCl, pH 8.2, 14 mM Mg(OAc)2, 60 mM KCl, 6 mM 2-mercaptoethanol). After washing, the pellet was suspended in 1.1 volumes of the pellet weight (mL = g) of S30 B buffer (10 mM Tris–HCl, pH 8.2, 14 mM Mg(OAc)2, 60 mM KOAc, 1 mM DTT and 1 × cOmplete protease inhibitor). Subsequently, the cells were lysed using a single French press passage at 1000 psig. The lysate was then centrifuged twice at 30,000×g, 4 °C for 30 min and the supernatant was subjected to a heat-step at 42 °C for 45 min. The lysate was then dialyzed in dialysis tubes of 10 MWCO against 5 L S30C buffer (10 mM Tris–acetate, pH 8.0, 14 mM Mg(OAc)2, 60 mM KOAc, 0.5 mM DTT) for 3 h at 4 °C and then for another 16 h at 4 °C in fresh 5 L S30C buffer. After dialysis, precipitate was removed by centrifugation at 30,000×g, 4 °C for 30 min.
Recombinant expression of branched chain amino acid aminotransferase IlvE
IlvE was expressed in E. coli BL21 Star express cells as described previously (Lazarova et al. 2018). Briefly, 100 mL LB medium with 100 µg/mL ampicillin in a 0.5 L baffled shaking flask was inoculated with a single colony of freshly transformed cells and incubated at 37 °C, 180 rpm for 16 h. Then 1 L LB medium with 100 µg/mL ampicillin in a 2 L baffled shaking flask was inoculated with 20 mL of the preculture and incubated at 37 °C, 180 rpm until OD600 0.6–0.9 was reached and 1 mM IPTG was added. The cells were cultivated for another 4 h under the same conditions before they were harvested, washed with IlvE buffer (100 mM Tris–HCl, pH 8.0, 100 mM NaCl), suspended in the same buffer with protease inhibitor cocktail and disrupted by sonication. The crude lysate was centrifuged at 30,000×g at 4 °C for 30 min, passed through a 0.45 µm filter and applied on a 5 mL HiTrap IMAC column previously equilibrated with IlvE buffer with 20 mM imidazole. The bound protein was then washed with another 15 CV of the same buffer and eluted with 4 CV IlvE buffer with 300 mM imidazole. The eluate was dialyzed against the 100-fold volume of IlvE buffer, concentrated using 10 kDa MWCO ultrafiltration devices and glycerol was added to a final concentration of 50% (v/v). The concentration of the protein stock was 10 mg/mL. The aliquoted protein solution was flash frozen in liquid nitrogen until further use.
Amino acid conversion of KIV and MOV
Conversion of KIV (3-methyl-13C, 3, 4, 4, 4-D4) to L-Val(4-13C, 2, 3, 4, 4, 4-D5) and MOV (4-methyl-13C, 3, 3, 4, 5, 5, 5-D6) to L-Leu (5-13C, 2, 3, 3, 4, 5, 5, 5-D7) was carried out in 10 mL conversion mixture and in D2O. Substance amounts to yield final concentrations of 100 mM for Tris and NaCl, 350 mM for L-Glu and 70 mM for KIV or MOV were weighed. The components were dissolved together in 9 mL D2O and pH was adjusted to 7.5 with 2 M NaOH in D2O. Then the mixtures were completed with 1 mL of 10 mg/mL IlvE solution to yield a final concentration of 1 mg/mL. The mixtures were incubated at 30 °C, 180 rpm for 16 h and passed through 10 kDa MWCO ultrafiltration devices to remove IlvE. The KIV or MOV permeates were then used instead of L-Val or L-Leu in CF expression reactions.
Cell-free protein expression
All proteins in this study were produced using S30 lysates prepared from E. coli K12 A19 and its indicated derivatives (Schwarz et al. 2007; Falkinham and Clark 1974). Briefly, expression was carried out in a two compartment CF expression configuration with a reaction mixture (RM) volume of 60 µL and a 13-fold feeding mixture (FM) volume. The compartments were separated by a 12–14 MWCO dialysis membrane. In a standard CF reaction, both compartments contained 1 mM of all 20 amino acids, an additional 1 mM each of L-Arg, L-Cys, L-Trp, L-Met, L-Asp and L-Glu, 20 mM acetylphosphate, 20 mM phosphoenolpyruvate, 2 mM dithiothreit, 0.21 mM folinic acid, 1 × cOmplete protease inhibitor, 100 mM HEPES–KOH, pH 8.0, 800 µM EDTA, 18 mM MgOAc and 170 mM KOAc. The RM additionally contained 0.3 U/µL RNasin, 0.5 mg/ml tRNA from E. coli MRE 600, 0.04 mg/ml pyruvate kinase, 0.35% (v/v) S30 lysate and 15 ng/µL template of either pET21a-GFP-6xHis, pIVEX2.3d-proteorhodopsin-6xHis or pIVEX2.3d-6xHis-CypD, respectively. Analytical scale screening reactions were conducted in customized reaction containers holding the RM and placed in cavities of 24-well microplates filled with 775 µl of FM. Expression of NMR samples was conducted with 3–6 mL RM and 15-fold FM in commercial Slide-A-Lyzer devices. Expression was carried out at 30 °C for 16 h at 180 rpm axial agitation. Subsequently, the RM was harvested and centrifuged at 18,000×g, 4 °C for 10 min.
Labeling and purification of his-tagged CypD
Truncated CypD (△43-207) was CF expressed from pIVEX2.1d-6xHis-CypD plasmid and IMAC purified via N-terminal 6xHis-tag as described previously (Hein et al. 2016). Briefly, 5 mL IMAC Sepharose 6 Fast Flow resin was used and purification was performed in 50 mM HEPES, pH 7.5, 150 mM NaCl with 20 mM imidazole in the washing buffer and 400 mM imidazole in the elution buffer. After elution, CypD was dialyzed against 50 mM sodium phosphate, pH 7.0, 0.5 mM DTT for 12–16 h and concentrated using 10 kDa MWCO ultrafiltration devices. Samples were supplemented with 100 µg/mL streptomycin, 1 × cOmplete protease inhibitor, 0.15 mM DSS and 5% D2O, and had a final protein concentration of 100–150 µM. Labeling was carried out using one of the 15N labeled amino acids L-Glu, L-Gln, L-Asp and L-Asn, respectively, while the other 19 amino acids were non-labeled. Inhibitors aminooxyacetate (AOA) and 6-diazo-5-oxo-norleucine (DON) were applied in some reactions at concentrations of 20 and 5 mM, respectively.
Labeling and purification of his-tagged proteorhodopsin
Proteorhodopsin (PR) was expressed and purified using a modified protocol previously published (Reckel et al. 2011). PR was CF expressed from pIVEX2.3d-proteorhodopsin-6×His plasmid in the presence of 0.4% (w/v) glyco-diosgenin (GDN) and 0.1% (w/v) diC7PC and purified via C-terminal 6×His-tag. PR methyl-labeled samples were CF synthesized in presence of 19 unlabeled amino acids and either 1 mM L-Val (4-13C, 2, 3, 4, 4, 4,-D5) converted from KIV (3-methyl-13C, 3, 4, 4, 4-D4) or 0.5 mM L-Leu (5-13C, 2, 3, 3, 4, 5, 5, 5-D7) converted from MOV (4-methyl-13C, 3, 3, 4, 5, 5, 5-D6). After harvesting, the samples were applied on an IMAC column equilibrated with NMR buffer (25 mM Na-acetate, pH 5.0, with 0.1% (w/v) diC7PC). The bound protein was then washed with 15 CV NMR buffer with 20 mM imidazole to remove impurities and GDN. The sample was eluted with NMR buffer containing 300 mM imidazole. The eluate was then concentrated to a volume of 1 mL using 10 MWCO centriprep ultrafiltration devices. The buffer was next exchanged on a PD Miditrap G25 column to NMR buffer without imidazole and concentrated again using 10 MWCO centriprep ultrafiltration devices. The final samples also contained 100 µg/mL streptomycin, 1 × complete protease inhibitor, 0.15 mM Sodium trimethylsilylpropanesulfonate and 5% D2O.
NMR measurements
All [15N, 1H]-TROSY spectra of CypD were acquired at sample temperatures of 303 K on Bruker 600 MHz (Avance II), 700 MHz (Avance III HD), 800 MHz (Avance III HD), 900 MHz (Avance Neo) and 950 MHz (Avance III) NMR spectrometers, equipped with cryogenic 1H triple-resonance probes. To take advantage of longitudinal 1H relaxation enhancement between scans, the Band-Selective Excitation Short-Transient (BEST) method was applied with proton pulses with a bandwidth of 4.8 ppm centered at 8.7 ppm (Farjon et al. 2009). The interscan delay was set to 0.3 s. All CypD samples had concentrations of 100–150 µM in a 5 mm NMR tube with 600 µL sample volumes.
Methyl 1H–13C correlation spectra of PR were acquired on a Bruker Avance Neo 950 MHz spectrometer equipped with a cryogenic probe at sample temperatures of 313 K. An XL-ALSOFAST-HMQC pulse sequence with the 13C–1H back-transfer period shortened to 2 ms combined with delayed decoupling was employed (Rößler et al. 2020a). The delay between scans was set to 0.7 s. Using gradient coherence selection, the sequence was suitable to eliminate otherwise strong t1-noise from detergent and acetate signals. The L-Val (4-13C, 2, 3, 4, 4, 4,-D5) labeled and the L-Leu (5-13C, 2, 3, 3, 4, 5, 5, 5-D7) labeled samples had concentrations of 80 µM and 120 µM, respectively, in 5 mm NMR tubes with 600 µL sample volume.


Comments (0)