
Remember me






The COVID-19 pandemic ravaging across the globe is caused by SARS-CoV-2 infection and manifests in humans as diverse signs and symptoms involving multiple organ systems, including cardiovascular, renal, and endocrine systems, as well as the brain. Initial reports documented at the onset of the pandemic focused primarily on the pulmonary pathophysiology of SARS-CoV-2 infections. However, with the progression of the pandemic, patient data providing evidence for the role of SARS-CoV-2 in eliciting damage to other organ systems began surfacing (Chakraborty & Basu, 2020). COVID-19 was observed to be accompanied by severe damage to the heart, characterized by myocardial and pericardial inflammation. Multiple instances of myocardial infarction were noted in patients with severe COVID-19. A reduction in cardiac ejection fraction was also frequently noted in patients with COVID-19. Renal complications associated with SARS-CoV-2 infection in patients culminated in mortality, highlighting the negative impact of coronavirus on diverse organ systems. Since both cardiomyocytes and kidney epithelial cells express angiotensin-converting enzyme-2 (ACE-2) protein on their surface and SARS-CoV-2 utilizes this protein to infect cells, direct infection of the aforementioned cell types by SARS-CoV-2 has been postulated to result in cardiac and renal pathology accompanying COVID-19. Indeed, evidence exists supporting the infection of cardiac muscle (Duarte-Neto et al., 2021) and renal tubular epithelial cells (Bradley et al., 2020) by SARS-CoV-2. In addition to the aforementioned organ systems that are affected by COVID-19, SARS-CoV-2 infection has been reported to severely perturb the physiology of other vital systems connecting every organ in the human body. Severe vascular dysfunction characterized by disseminated clotting across vascular beds involving multiple organs has been associated with COVID-19 (Varga et al., 2020). Further, altered vascular permeability and enhanced immune cell trafficking into tissue parenchyma contribute significantly to perturbed homeostasis in SARS-CoV-2 infection. Although an exaggerated immune response to viral infection is thought to underscore abnormalities in clotting, the direct involvement of endothelial cells activated as a result of viral infection or immune responses has also been associated with clotting abnormalities. Inappropriate activation of the clotting cascade has been observed across pulmonary vascular beds in individuals with SARS-CoV-2 infection, thus resulting in pathology (Bradley et al., 2020). In addition, autopsy reports have revealed the prevalence of capillary microthrombi in extra-pulmonary sites (Varga et al., 2020). Owing to ACE-2 expression by vascular endothelial cells, SARS-CoV-2 infection mediated by ACE-2 can result in endothelial cell death via apoptosis or pyroptosis, leading to disruptions in blood vessel wall integrity and altered vascular permeability. Virus-induced changes in vascular physiology affect multiple organ systems, resulting in the constellation of symptoms characteristic of COVID-19.
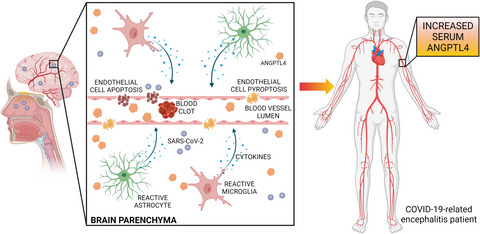
Reports of patients with COVID-19 exhibiting neurological symptoms indicating the involvement of the brain have emphasized the potential of CNS infection by SARS-CoV-2 (Ellul et al., 2020). Extant evidence supports the hypothesis that the nasal epithelium acts as a conduit for viral entry into the CNS, as demonstrated by the presence of SARS-CoV-2 infection in olfactory epithelium supporting cells, including sustentacular and horizontal basal cells (Brann et al., 2020). Olfactory sensory neurons have not been observed to be infected by SARS-CoV-2. However, a recent study revealed SARS-CoV-2 immunostaining in olfactory mucosa sensory neurons and connected brain regions, implicating olfactory sensory nerve endings in virus egress from the periphery to CNS (Meinhardt et al., 2021). Patients reported symptoms including impaired consciousness, confusion, delirium, agitation, psychosis, and neurocognitive (dementia-like) syndrome. Imaging studies employing magnetic resonance imaging (MRI) have revealed hyperintense signals in multiple brain regions, including the temporal lobe, hippocampus, ventricular walls, leptomeningeal spaces, and diffuse white matter abnormalities. Electroencephalography (EEG) findings in the majority of patients have revealed non-specific slow-wave activity without any epileptic patterns. The absence of the virus in the cerebrospinal fluid (CSF) of patients with COVID-19-related encephalitis indicates that these neurological insults may be independent of direct viral action upon neurons. Indeed, electron microscopy analyses have demonstrated the absence of SARS-CoV-2 in neurons (Paniz-Mondolfi et al., 2020). Examination of autopsied brain sections of patients with COVID-19-related encephalitis has revealed both focal and diffuse lesions attributed to an excessive inflammatory response and hemorrhage. Both astrocyte and microglial activation accompany inflammation of brain parenchyma as a result of CNS infection by SARS-CoV-2 (Solomon, 2021). The distribution of activated astrocytes across the entire brain is generally diffuse, but microglial activity is observed predominantly in the cerebellum and brainstem. Cytotoxic T-cells (Tc-cells) have also been reported to patrol the brainstem and meninges of patients. Diffuse perivascular leucocytic infiltrates have been observed, signifying the magnitude of proinflammatory responses elicited in the brain as a result of viral entry. In addition to the lesions associated with overt inflammatory reactions, considerable evidence points to the role of acute cerebrovascular injury and ischemia in the development of neurological symptoms in COVID-19-related encephalitis. Loss of cerebrovascular integrity as a result of brain-vascular endothelial cell activation/viral infection may result in cerebral hemorrhage that varies in magnitude depending upon the extent of vascular damage. Hypercoagulability-induced cerebral blood vessel occlusion can culminate in stroke, thus contributing to ischemic brain lesions observed in patients with COVID-19-associated encephalitis. This evidence highlights the potential role played by cerebral vasculature damage that is either elicited by a viral infection or an altered coagulative state alongside hyperactive immune responses that contribute to encephalopathy upon SARS-CoV-2 infection (Figure 1).

Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) entry into the central nervous system (CNS) via the cribriform plate from olfactory mucosa is associated with the activation of brain-resident innate immune cells such as astrocytes and microglia. These activated cells secrete proinflammatory cytokines and chemokines which activate endothelial cells, hence modulating cerebrovascular permeability and blood coagulability. Enhanced thrombosis in cerebral vasculature has been shown to cause blood vessel occlusion followed by ischemic injury to the brain. In addition, direct infection of vascular endothelial cells by SARS-CoV-2 results in cell death by apoptosis or pyroptosis, thus further degrading vascular integrity. Hypoxia-induced angiopoietin-like 4 (ANGPTL4) up-regulation in brain parenchyma is released into the systemic circulation, thus increasing ANGPTL4 abundance in the serum and indicating the onset/progression of coronavirus disease-19 (COVID-19)-related encephalitis
1 ANGPTL4 AS A BIOMARKER FOR COVID-19-ASSOCIATED ENCEPHALITISThe reported findings discussed above (Altmayer et al., 2021) regarding the neurological complications associated with COVID-19 warrant further investigations to decipher the mechanistic basis of CNS damage. This will facilitate the development of potential therapeutic strategies, thus reducing disease morbidity. In addition to elucidating the molecular events underpinning CNS injury, the discovery of molecular markers associated with SARS-CoV-2-associated encephalitis may help physicians to initiate therapy aimed at controlling CNS damage, thus curtailing further progression and ameliorating disease burden. In an attempt to discover biomarkers associated with COVID-19-associated encephalitis, the report by Altmayer et al. demonstrated an up-regulation of serum ANGPTL4 in patients with COVID-19-related encephalitis compared to patients without CNS inflammation. The authors classified COVID-19 patients into two groups (control or encephalitis groups) based on the presence/absence of neurological manifestations as determined by findings from routine neurological examinations (characterized by impaired cognition, delirium, seizure, and confusion), EEG, and MRI. COVID-19 patients exhibiting abnormalities in EEG, MRI findings, or neurological deficits identified in routine neurological examinations were included in the encephalitis group. In contrast, patients presenting with no neurological impairments were included in the control group. Estimation of molecular markers of endothelial damage in serum revealed higher endothelial dysfunction in the encephalitis group than in the control group. E-selectin and thrombomodulin are molecules expressed by endothelial cells that are involved in leukocyte adhesion to endothelium and inhibition of the coagulation cascade overactivation, respectively. Damage to vascular endothelial cells causes the release of E-selectin and thrombomodulin into the circulation, thereby increasing their abundance in serum. Soluble E-selectin and thrombomodulin were observed to be up-regulated in the serum of patients in the encephalitis group. Serum ‘a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13’ (ADAMTS13) activity was demonstrated to be reduced in the COVID-19-associated encephalitis group compared to that in the non-encephalitis group. ADAMTS13 activity to Von Willebrand Factor A2 domain (vWF-A2) ratio in serum was observed to be lower in the encephalitis group than in the control group, indicating an increased propensity for microvascular thrombosis in patients with encephalitis. In addition to these findings denoting greater endothelial damage in COVID-19-associated encephalitis, the abundance of proinflammatory cytokines such as tumor necrosis factor-α (TNF- and interleukin-6 (IL-6) were demonstrated to be up-regulated in the serum of patients exhibiting neurological manifestations. A positive correlation of serum TNF-
and interleukin-6 (IL-6) were demonstrated to be up-regulated in the serum of patients exhibiting neurological manifestations. A positive correlation of serum TNF- with soluble E-selectin and thrombomodulin abundance highlights, but does not conclusively establish, the involvement of an exaggerated immune response in eliciting cerebrovascular endothelial cell activation and dysfunction following SARS-CoV-2 infection, thus promoting COVID-19-related encephalopathy. Serum ANGPTL4 abundance, which was up-regulated in the encephalitis group compared to that in the control group, lacked a correlation with TNF-α levels in patients in the encephalitis group or in the state of hypoxia related to acute respiratory distress syndrome (ARDS) in patients (PaO2/FiO2 ratio). These findings excluded a relationship of serum ANGPTL4 abundance with systemic immune responses or differential effects of hypoxemia in the control versus encephalitis groups; rather, they reinforce a direct association of ANGPTL4 abundance with CNS injury because of CNS infection by SARS-CoV-2. The positive correlation of systemic inflammation with SARS-CoV-2-associated encephalitis but not with ANGPTL4 seems contradictory, especially given that ANGPTL4 is stated as a biomarker associated with COVID-19-related encephalitis. However, the migration of viruses into the CNS via the cribriform plate from the olfactory epithelium may lead to CNS inflammation characterized by activated astrocytes and microglia. Secretion of proinflammatory cytokines by activated inflammatory cells into the brain parenchyma can result in cerebrovascular coagulopathy and endothelial cell activation, thus causing cerebral ischemia and altered vascular permeability, respectively. Upon entering the brain, SARS-CoV-2 is capable of infecting vascular endothelial cells, which promotes their death and culminates in microhemorrhages. Ischemic brain injury upregulates ANGPTL4 secretion by cells which then escapes into the systemic circulation owing to the disintegration of cerebral vasculature as a consequence of neuroinflammation and a viral insult to endothelial cells, which may indicate the progression of SARS-CoV-2-associated encephalitis. Conversely, ANGPTL4 up-regulation, rather than being a consequence of COVID-19-related encephalitis, may act as a causative agent in the pathogenesis of COVID-19-related encephalitis. Indeed, existing reports indicate a role of ANGPTL4 in promoting vascular permeability by destabilizing endothelial cell junctions (Guo et al., 2014). In this regard, further studies investigating the mechanistic basis of ANGPTL4 up-regulation may reveal future therapeutic strategies for preventing COVID-19-related encephalitis. COVID-19-associated encephalitis in recovered patients has been shown to be accompanied by similar levels of mortality during hospital stay. Although the data documented in the report provide significant evidence supporting ANGPTL4 as a molecular marker for COVID-19-associated encephalitis, besides being limited by its sample size, another drawback of the study is that serum ANGPTL4 levels were measured only once after patients were admitted to the intensive care unit (ICU) following diagnosis of COVID-related encephalitis. A prospective cohort study estimating serum ANGPTL4 in COVID-19 patients at multiple time-points following admission and recording the incidence of encephalitis development and disability scores associated with encephalitis may verify the potential of ANGPTL4 as a predictive and diagnostic tool as well as a prognostic biomarker of SARS-CoV-2-related encephalitis.
with soluble E-selectin and thrombomodulin abundance highlights, but does not conclusively establish, the involvement of an exaggerated immune response in eliciting cerebrovascular endothelial cell activation and dysfunction following SARS-CoV-2 infection, thus promoting COVID-19-related encephalopathy. Serum ANGPTL4 abundance, which was up-regulated in the encephalitis group compared to that in the control group, lacked a correlation with TNF-α levels in patients in the encephalitis group or in the state of hypoxia related to acute respiratory distress syndrome (ARDS) in patients (PaO2/FiO2 ratio). These findings excluded a relationship of serum ANGPTL4 abundance with systemic immune responses or differential effects of hypoxemia in the control versus encephalitis groups; rather, they reinforce a direct association of ANGPTL4 abundance with CNS injury because of CNS infection by SARS-CoV-2. The positive correlation of systemic inflammation with SARS-CoV-2-associated encephalitis but not with ANGPTL4 seems contradictory, especially given that ANGPTL4 is stated as a biomarker associated with COVID-19-related encephalitis. However, the migration of viruses into the CNS via the cribriform plate from the olfactory epithelium may lead to CNS inflammation characterized by activated astrocytes and microglia. Secretion of proinflammatory cytokines by activated inflammatory cells into the brain parenchyma can result in cerebrovascular coagulopathy and endothelial cell activation, thus causing cerebral ischemia and altered vascular permeability, respectively. Upon entering the brain, SARS-CoV-2 is capable of infecting vascular endothelial cells, which promotes their death and culminates in microhemorrhages. Ischemic brain injury upregulates ANGPTL4 secretion by cells which then escapes into the systemic circulation owing to the disintegration of cerebral vasculature as a consequence of neuroinflammation and a viral insult to endothelial cells, which may indicate the progression of SARS-CoV-2-associated encephalitis. Conversely, ANGPTL4 up-regulation, rather than being a consequence of COVID-19-related encephalitis, may act as a causative agent in the pathogenesis of COVID-19-related encephalitis. Indeed, existing reports indicate a role of ANGPTL4 in promoting vascular permeability by destabilizing endothelial cell junctions (Guo et al., 2014). In this regard, further studies investigating the mechanistic basis of ANGPTL4 up-regulation may reveal future therapeutic strategies for preventing COVID-19-related encephalitis. COVID-19-associated encephalitis in recovered patients has been shown to be accompanied by similar levels of mortality during hospital stay. Although the data documented in the report provide significant evidence supporting ANGPTL4 as a molecular marker for COVID-19-associated encephalitis, besides being limited by its sample size, another drawback of the study is that serum ANGPTL4 levels were measured only once after patients were admitted to the intensive care unit (ICU) following diagnosis of COVID-related encephalitis. A prospective cohort study estimating serum ANGPTL4 in COVID-19 patients at multiple time-points following admission and recording the incidence of encephalitis development and disability scores associated with encephalitis may verify the potential of ANGPTL4 as a predictive and diagnostic tool as well as a prognostic biomarker of SARS-CoV-2-related encephalitis.
Anirban Basu is a recipient of a J C Bose Fellowship (JCB/2020/000037) from the Science and Engineering Research Board (SERB). The image was prepared using Biorender (https://biorender.com). Victor Villemagne, Sarah Ch’ng and Laura Hausmann revised the text as part of Journal of Neurochemistry’s Editorial team.
CONFLICT OF INTERESTAnirban Basu is an Editor for the Journal of Neurochemistry. The authors declare no further potential conflict of interest.
AUTHOR CONTRIBUTIONSBoth authors drafted the text, and approved of its final version.
Comments (0)