Tuning the quasi‐harmonic treatment of crystalline ionic liquids within the density functional theory
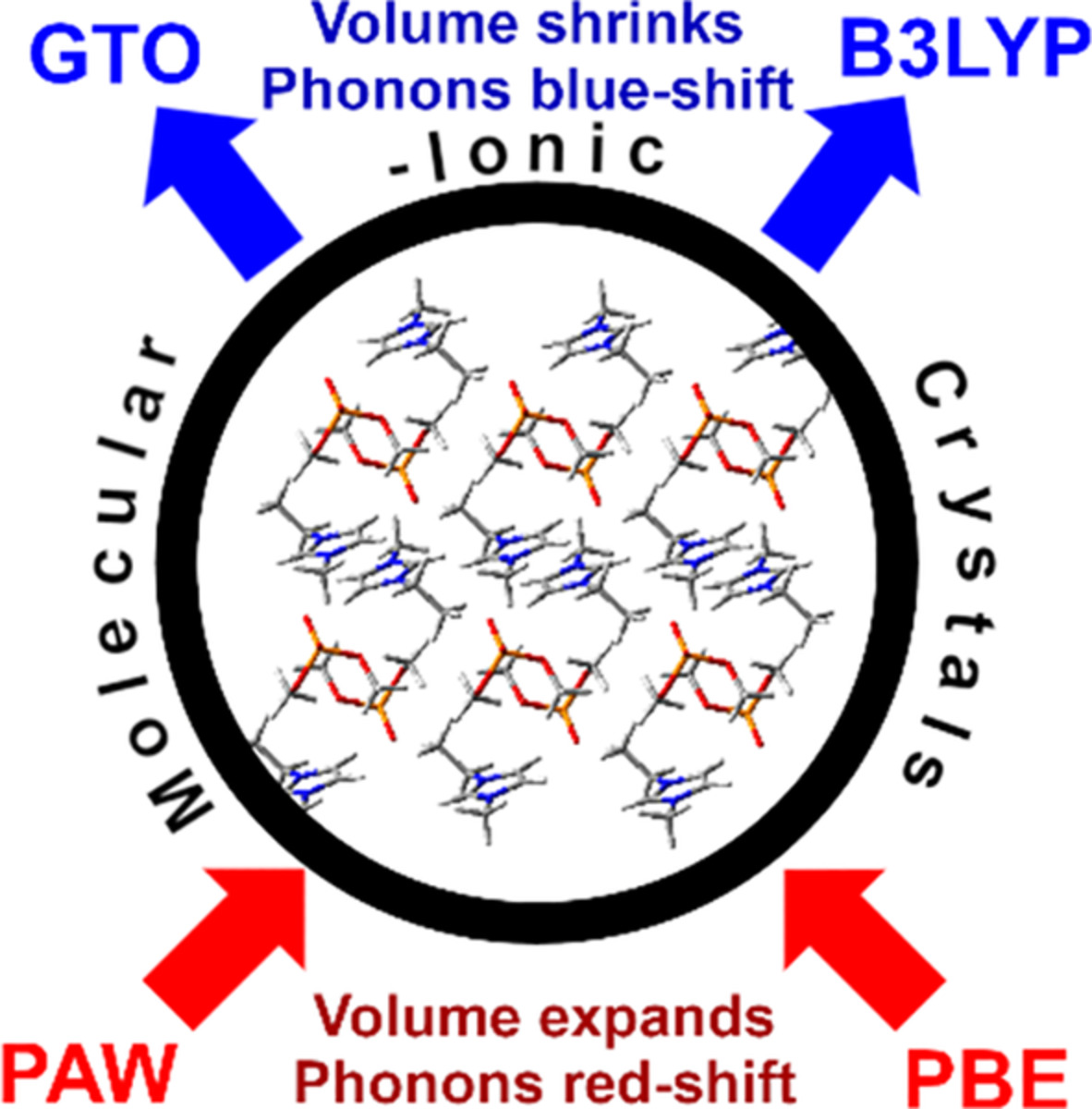
Five ionic liquids are selected for benchmarking the performance of quasi-harmonic density functional theory (DFT) calculations of structural, phonon, and thermodynamic properties of their crystals. Data predicted by individual computational setups are sorted, establishing a distinct hierarchy among the first-principles approaches. PBE-D3 and B3LYP-D3 functionals are coupled with various plane wave and Gaussian-type orbital (GTO) basis sets. Propagation of the basis set superposition error and of the imperfections of both functionals into finite-temperature properties is discussed in detail. PBE-D3 together with a triple-zeta GTO basis set often yields the most accurate predictions of predicted molar volume and heat capacity with errors at 1% and 8%, respectively, representing the state-of-the-art for quasi-harmonic DFT calculations for crystalline ionic liquids. Fortuitous error cancellation between the basis-set superposition (overbinding) and PBE imperfection (overexpanding) strongly affects the overall accuracy, unlike the case of B3LYP/GTO calculations, impeding systematic convergence of the methodology towards higher accuracy.



Comments (0)