
Remember me


Lignin is one of the three main structural components that is essential to all biomass.1, 2 Together with the other two components—cellulose and hemicellulose—lignin is one of the most abundant polymers worldwide. Today, the major part of lignin produced is Kraft lignin; it is a by-product of the Kraft pulping process used in pulp-and-paper mills worldwide.3 There, the Kraft lignin is mostly burned to keep the costs of the pulping process low.4 However, more value-added lignin applications are on the horizon; either lignin is to be broken down into small molecule building blocks for fine-chemicals5 or it can be used whole in novel polymer composites.6 Several of these lignin-polymer composites are potential precursors to highly valuable carbon fibers (CF).7-12 The approach of incorporating carbon nanotubes (CNT) into the composite precursor is highly promising.13
Lignin is a three-dimensional network polymer composed of mainly three phenylpropanoid building blocks which are shown in Figure 1: p-coumaryl alcohol, coniferyl alcohol, and syringyl alcohol.14 The actual polymeric structure of native lignin varies between and within plants. In lignins extracted from plants, the structure depends also strongly on the method of isolation.15

The three basic monolignols (from left to right): p-coumaryl alcohol, coniferyl alcohol, and syringyl alcohol
CNT are defined as slim tubes made of sp2-hybridized carbon atoms with a nanoscale diameter and an enormous aspect ratio.16 CNT appear in three different helicities: “armchair,” “zigzag,” or “chiral.”17 The helicity strongly influences its electronic properties—the CNT might be metallic or a semiconductor with either a narrow or a wide band-gap.18 Since the initial discovery of CNT19 and their belated increase in popularity,20 many applications in either mechanical material reinforcement21 or conducting22 and storing electric energy23 have been developed.
Composites from lignin and CNT are promising precursors to carbon fibers.13 In lignin-CNT composites, the precise control over the lignin-CNT interface remains an open issue,7, 24, 25 which warrants a fundamental investigation into the noncovalent interactions involved.
Noncovalent interactions are a lot less clear defined than their covalent counterparts.26 Noncovalent interactions appear between partners that are separated at the 10–100 Å distance scale.27, 28 In contrast, covalent interactions take place at 2–4 Å distance.26 While being weaker in strength than their covalent counterparts, noncovalent interactions influence and shape our physical world: for example, “the very existence of a liquid phase, and also all related effects like solvation phenomena, can be attributed to non-covalent interactions.”29 Non covalent interactions also play a key role in biology, where they for example direct the binding of antibodies or dictate the structure of our very DNA.30, 31 Other self-assembly processes, for example, in the solid phase, are caused by noncovalent interactions too.32
On a fundamental level, Israelachvili distinguishes noncovalent interactions into classes such as interactions involving polar molecules, interactions involving the polarization of molecules, van der Waals forces (which subconsists of dispersion−/London-, induction-, and orientation forces) and hydrogen bonding.33 Chemically more intuitive categorizations refer to the dominant types of interacting atomic orbitals, such as π-stacking,26 CHπ and OHπ interaction.34 The latter two noncovalent interactions are also commonly subgrouped into the hydrogen bonding category35—a category that is as chemically important36 as it is hard to define conceptually.37, 38 Also other, more detailed categorizations of π interactions appeared in the literature about dispersive interactions of small-molecule model compounds.39-43
Computational studies are in common in CNT-related research, as the following selection of literature examples is supposed to demonstrate. Charlier's review on defects in CNT for example is mostly comprised of references to computational works.44 Tournous and Charlier also published a density functional theory local density approximation (DFT-LDA) study of benzene adsorbing on finite single-walled CNT (SWCNT) models.45 Andzelm et al. researched the simulated adsorption of ammonium over intact and defective SWCNT.46 Amorim et al. calculated the stability of divacancy defects in SWCNT,47 as did Berber and Oshiyama.48 Azadi et al. simulated the effect differently oriented Stone–Wales defects in finite SWCNT models.49 Lopez-Bezanilla et al. used the first-principles local-orbital density functional method to investigate the charge transport properties of functionalized CNT.50, 51 Tkatchenko et al. found that applying DFT to supramolecular host–guest systems requires and efficient inclusion of dispersion effects.52
In lignin-related research, computational studies are not quite as common—some notable instances are mentioned in the following lines. Quite early, Glasser and Glasser used the SIMREL program to simulate the formation of a dehydrogenation polymer of coniferyl alcohol starting from monomers—contrary to the literature mentioned so far, the method is not electron-based but rather relies on abstractions of the chemical formula.53 Barsberg et al. on the other hand conducted and reported a succession of papers, in which they simulate Raman-spectra of lignin model compounds to compare them to experimental values.54-56 Usually though, polymeric and monomeric lignin models are investigated by chemical experiment.57
The noncovalent interactions between lignin and CNT have only—to the best of our knowledge—been addressed on an ab-initio basis in our recent publication, where we investigated the influence of CNT-oxidation on the CNT-lignin interaction potential.58 In the previous study, we found that oxidation enables and enhances the interaction between CNT and organosolv lignin; we were able to simulate this finding using DFT-based adsorption simulations.
In this article, we further investigate the lignin-CNT interaction potential toward the quality and quantity of the occurring noncovalent interactions. We develop appropriate simplified model structures to represent lignin structural motifs and finally ask the question “How do SWCNT interact with cyclic small probe molecules? How strong are the resulting noncovalent interactions? What is the impact of the SWCNT diameter on the noncovalent interaction strength?”
2 METHODS 2.1 Computational setupWe carry out DFT simulations to calculate electron-density dependent ground state energies and structures of model adsorption complexes as implemented in the GPAW package.59 GPAW uses Blöchl's projector augmented wave,60 where the smooth part of the electron density and Kohn–Sham states is realized on three-dimensional real-space grids.61 This allows to set a variety of boundary conditions—for example, only one of three special dimension to be infinite. Additionally, the grid-based approach prevents basis-set superposition errors and enables a straightforward interpolation to the complete basis set limit.62
We approximate the exchange correlation energy in the generalized gradient approximation (GGA) as devised by Perdew, Burke, and Ernzerhoff (PBE)63:
Noncovalent interactions appear at longer distance scales than GGA are designed to handle. Especially London-like dispersion effects can notoriously not be accounted for by GGA.29, 52 Many dispersion correction approaches64-66 add the missing dispersion interaction to the GGA energy (1)
(1)
 (2)assuming that the GGA energy
(2)assuming that the GGA energy  does not contain any long range dispersion contribution at all. We adopt this strategy and use the almost parameter-free method proposed by Tkatchenko and Scheffler (TS09)65 to evaluate the dispersion contribution
does not contain any long range dispersion contribution at all. We adopt this strategy and use the almost parameter-free method proposed by Tkatchenko and Scheffler (TS09)65 to evaluate the dispersion contribution  .
Our simulations are set up in a way that the adsorption energy
.
Our simulations are set up in a way that the adsorption energy  is fully noncovalent, that is, no chemical bonds are established between CNTs and lignin model molecules.
is fully noncovalent, that is, no chemical bonds are established between CNTs and lignin model molecules.  is defined as the difference between the total energies of the adsorption complex
is defined as the difference between the total energies of the adsorption complex  and the sum of the individual reactants' total energies
and the sum of the individual reactants' total energies  and
and  calculated at the TS09-level58
calculated at the TS09-level58
 (3)thus yielding negative values in case of attractive interactions.
Conceptually, the total noncovalent interaction energy
(3)thus yielding negative values in case of attractive interactions.
Conceptually, the total noncovalent interaction energy  may be split into the two main fundamental contributions.34 These are (1) exchange-repulsion, electrostatic interactions, polarization, charge transfer and (2) dispersive interactions covered by the TS09 correction.67 We name the first contribution
may be split into the two main fundamental contributions.34 These are (1) exchange-repulsion, electrostatic interactions, polarization, charge transfer and (2) dispersive interactions covered by the TS09 correction.67 We name the first contribution  that is
that is
 (4)as obtained from the PBE energies
(4)as obtained from the PBE energies  evaluated at the TS09 relaxed geometries. This allows us to extract an approximate dispersive contribution through
evaluated at the TS09 relaxed geometries. This allows us to extract an approximate dispersive contribution through
 (5)
2.2 Structural models
(5)
2.2 Structural models
All structural models were set up using the atomic simulation environment ASE68, 69 while the subsequent output was analyzed with Avogadro.69, 70
The supercell—in which the structures were set up—measured 48 Å in both x and y direction. In the z-direction, the cell had the exact length of 5 CNT repetition units as set up by ASE (~12.3 Å).69 The CNTs were aligned along the z axis, consequently periodic boundary conditions were applied in this direction. Dirichlet boundary conditions (the electron density is set to zero at the boundary) were applied to the perpendicular x and y directions. The simulation grid was composed of 256 grid points both x and y directions as well as 64 grid points in z direction. The structures were optimized until a force gradient of 0.02 eV/Å was reached.
We restricted our study to single wall carbon nanotubes (SWCNT) of three different diameters, that is, (4,4), (8,8) and (12,12)-SWCNT (cf. Figures S1 and 2A)—as models for the CNTs. Due to their (n, n)-chirality, the SWCNT are considered all metallic and conducting.71 The unit cell contained five repetition units aligned along the z axis. As our base model, we considered our SWCNT to exhibit a pristine surface (p-SWCNT)—similar to the studies of Tournous and Charlier.45

(A) p-SWCNT of (4,4), (8,8) and (12,12) size (from left or right). (B) A chiral Stone–Wales defect—Highlighted in blue—In a (8,8)-sw-SWCNT segment as seen from the z-axis. Note that the perspective reveals the defect-induced deformation. (C) Close-up of the functional OH-group on a (12,12)-o-SWCNT segment
We also took into account the appearance of two type of defects. The first one is the Stone–Wales (SW) defects that occur in extended sp2-hybridized carbon structures when a single σCCbond is rotated by 90°.72 Up to 5% of all carbon atoms as-prepared SWCNT contain defects, which are most likely SW-defects.73 There are either straight or chiral SW defects46 in CNT.
Similar to the model of Azadi et al.49 we considered chiral SW defects (sw-SWCNT), as illustrated in Figure 2B.
The second defect considered serves as a model for oxidized CNTs. Here the nanotube was functionalized with a hydroxyl-group (o-SWCNT), similar to the models of Milowska and Majewski74 as depicted in Figure 2C.
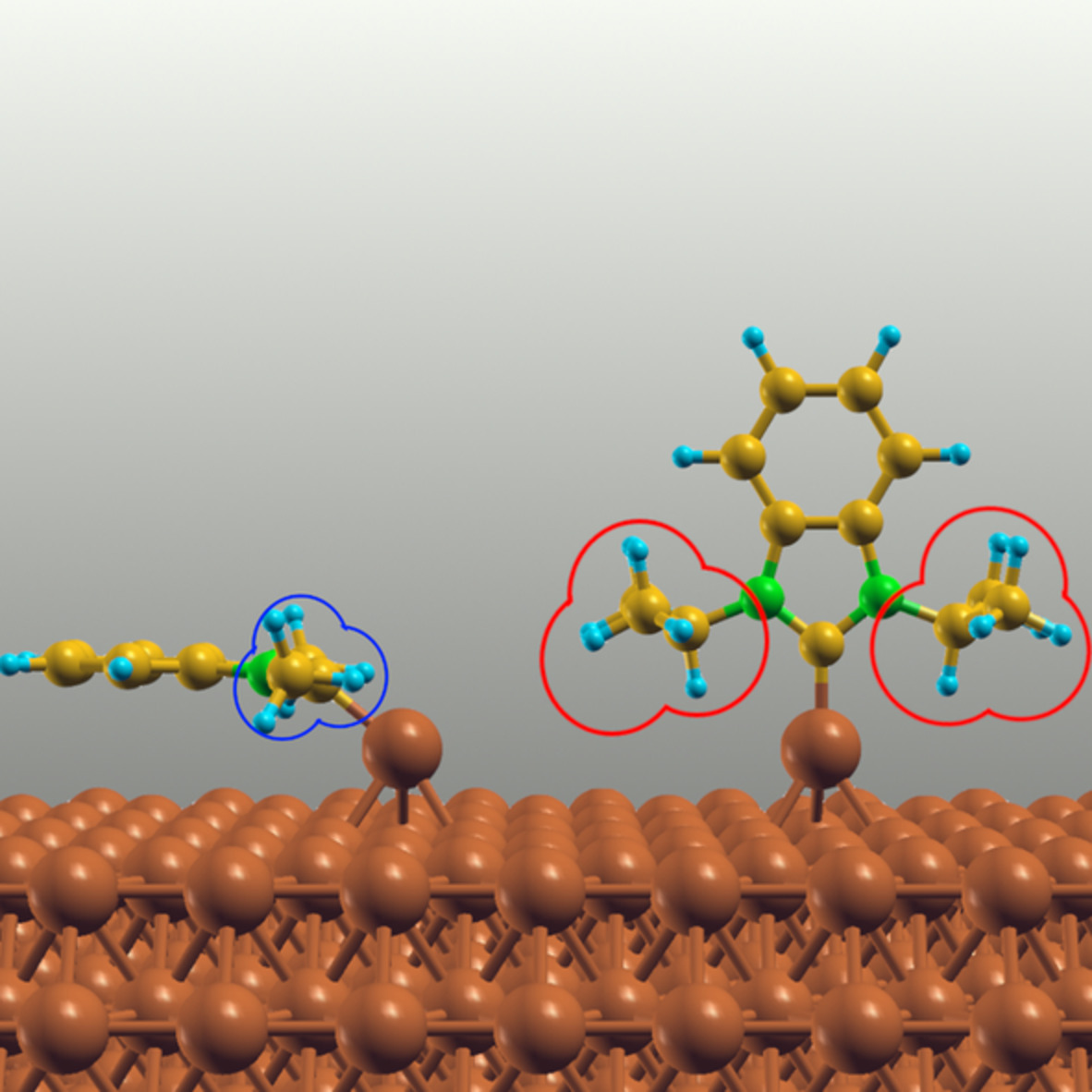
In order to model the interaction to lignin with its large variety of comprising functional parts, we selected several small molecules to interact with the SWCNTs. These are depicted in Figure 3A–F: cyclohexane, cyclohexanol and methoxycyclohexane represent the sp3-hybridized carbons and the aliphatic hydroxy- and methoxy-groups present in lignin. In addition, benzene, phenol, and anisol represent sp2-hybridized carbons and the aromatic functionalities of lignin.

Left: Schematic comparison of a monolignol randomly incorporated into lignin. Right: The model molecules chosen to represent important lignin-CNT interactions—(A) cyclohexane, (B) cyclohexanol, (C) methoxycyclohexane, (D) benzene, (E) phenol, and (F) anisol
The nature of the noncovalent interaction involved is not only determined by the nature of the adsorbing molecules themselves. Another important factor is the adsorbing molecules' orientation relative to the SWCNT. Weak forces often do not restrict the relative orientation of the constituents, but allow for many different local minima. In our adsorption calculations, the adsorbing molecules are oriented in up to four distinct positions (cf. Figure 4): The adsorbing molecule can either be oriented parallel to the SWCNT's surface—this is the p-position. The adsorbing cyclic molecule can also be aligned that the edge created by the β- and γ-standing carbon units is aligned with the SWCNT surface while all other carbon units are as far away from the SWCNT surface as possible—this is the e-position. The adsorbing molecule could also be oriented in a way, that either the α- or δ-standing carbon units point straight down to the SWCNT surface—these positions are called the α- or δ-position respectively.

The nature of the interaction between phenol and a (12,12)-SWCNT is controlled by the phenol's angle of approach. From left to right: Parallel (p-), edge (e-), α-, and δ- modes of adsorption
3 RESULTS 3.1 Defect formation in SWCNT We first discuss the energy needed to form the defects in the CNTs. The formation energies of both straight and chiral SW defects depend on CNT curvature C—the inverse of the nanotubes' radius r (6)
(6)
In agreement with the referenced data of Azadi et al.49 and Saidi et al.,75 our results indicate that chiral SW defect formation energies decrease with increasing curvature (cf. Figure 5). Our SW defects exhibit a positive, that is, unfavorable formation energy since the induce deformation to the host CNT77 (see also Figure 2).
 The formation energies of defective SWCNT depend on the SWCNTs' curvature (defined in Equation 6). OH-related defects are marked with circles, parallel SW-defects are marked by squares and chiral SW-defects are marked by diamonds. References for oxidized graphene from Hassan et al.76 as well as for series of chiral SW defects from Azadi et al.49 and Saidi et al.75 are also given
The formation energies of defective SWCNT depend on the SWCNTs' curvature (defined in Equation 6). OH-related defects are marked with circles, parallel SW-defects are marked by squares and chiral SW-defects are marked by diamonds. References for oxidized graphene from Hassan et al.76 as well as for series of chiral SW defects from Azadi et al.49 and Saidi et al.75 are also given
The binding of an OH group in o-SWCNT (cf. Figure 5) is found with negative  (i.e., favorable binding) and further decreases with increasing SWCNT-curvature. This dependence on C is understandable as the formation of the defect rehybridizes the C atom to which it is attached. This in turn releases curvature-induced stress—hence the more negative
(i.e., favorable binding) and further decreases with increasing SWCNT-curvature. This dependence on C is understandable as the formation of the defect rehybridizes the C atom to which it is attached. This in turn releases curvature-induced stress—hence the more negative  at higher curvature (i.e., lower radii). The extrapolation of our data to reference systems from literature (here: oxidized graphene76) is consistent.
at higher curvature (i.e., lower radii). The extrapolation of our data to reference systems from literature (here: oxidized graphene76) is consistent.
We now discuss, how the adsorbing molecules' substituents influence the binding energies with the nanotubes. Figure 6 shows an overview of the interaction energies between pristine SWCNT and the selection of model molecules from Figure 3. There,  and
and  denote the aliphatic and aromatic substituent respectively. A similar plot which includes also the respective
denote the aliphatic and aromatic substituent respectively. A similar plot which includes also the respective  for (4,4) and (8,8) p-SWCNT is given in Figure S4, where the same trends can be observed.
for (4,4) and (8,8) p-SWCNT is given in Figure S4, where the same trends can be observed.
 Adsorption of probe molecules onto (12,12) p-SWCNT. The lines connect the data points to allow for easier visual orientation—They do not embody any numerical meaning by themselves. A similar plot which includes also the respective
Adsorption of probe molecules onto (12,12) p-SWCNT. The lines connect the data points to allow for easier visual orientation—They do not embody any numerical meaning by themselves. A similar plot which includes also the respective  for (4,4) and (8,8) p-SWCNT is given in Figure S4. (All connecting lines are just given as visual guidance)
for (4,4) and (8,8) p-SWCNT is given in Figure S4. (All connecting lines are just given as visual guidance)
In general, the variation in  is dominated by
is dominated by  , whose variations are larger in magnitude than those of
, whose variations are larger in magnitude than those of  . No clear trends can be found for
. No clear trends can be found for  while the following observations are true for both
while the following observations are true for both  and
and  .
.
Focusing on  , the parallel configuration (p) leads to the strongest interaction in all the molecules, which is due to maximal interaction area in this orientation. Decreasing the interaction area—by enforcing any other configuration—strongly reduces the interaction. Within these configurations, the edge (e) orientation has still a slightly larger interaction area than a and δ and hence shows increased interaction with the CNT. Similarly, Gung et al. observed that relative substituent orientation strongly influenced intramolecular stacking interaction strengths in model compounds derived from 1,9-disubstituted triptycene.78
, the parallel configuration (p) leads to the strongest interaction in all the molecules, which is due to maximal interaction area in this orientation. Decreasing the interaction area—by enforcing any other configuration—strongly reduces the interaction. Within these configurations, the edge (e) orientation has still a slightly larger interaction area than a and δ and hence shows increased interaction with the CNT. Similarly, Gung et al. observed that relative substituent orientation strongly influenced intramolecular stacking interaction strengths in model compounds derived from 1,9-disubstituted triptycene.78
There is not much difference in the attractive forces of aliphatic and aromatic compounds, which is somehow unexpected, but was found similarly for bound pairs of small molecules.79
Interestingly, there is also a slight trend to higher binding energies with increasing size of  and
and  . We could not relate this effect to the Hammett parameter of the substituents as proposed by Wheeler and Houk,80 as neither the meta parameter
. We could not relate this effect to the Hammett parameter of the substituents as proposed by Wheeler and Houk,80 as neither the meta parameter  nor the para parameter
nor the para parameter  increase in H, OH, OMe.81, 82 The interaction is rather a local—interaction with the substituent41, 83 and its strength is connected to the size of the substituent—therefore can be regarded as geometric effect. This is in line with the observation that adsorption interaction of substituted benzene dimers was enhanced regardless of electron withdrawing and donating groups, as the substituent groups also tend to increase interaction.42
increase in H, OH, OMe.81, 82 The interaction is rather a local—interaction with the substituent41, 83 and its strength is connected to the size of the substituent—therefore can be regarded as geometric effect. This is in line with the observation that adsorption interaction of substituted benzene dimers was enhanced regardless of electron withdrawing and donating groups, as the substituent groups also tend to increase interaction.42
We interpret adsorptions in the δ-position as an indicator of the interaction potential of the individual CH2- (e.g., cyclohexane-δ) or CH- (benzene-δ) group. With all substituents being equal, individual CH2 groups adsorb slightly stronger on the p-SWCNTs than individual CH groups. This could be tied to the comparably larger area of interaction (two hydrogen atoms on the surface proximity vs. one hydrogen atom) that the individual CH2 assumes. Electron donating hydroxy- and methoxy groups slightly enhance the δ-adsorption of CH2 groups in the cyclic adsorbants.
Figure S5 demonstrates that sw-defects do not influence the adsorption of benzene on (4,4), (8,8), or (12,12) SWCNT significantly in either  ,
,  , or
, or  .
.
In the following, we further analyze the interactions observed in regards to their interaction class: The first class is the ππ interaction26, 79—sometimes referred to as π-stacking.84, 85 The second interaction class consists of three types of hydrogen bonding. Hydrogen bonding is a broad interaction class.86 It occurs, for example, between water molecules (or other molecules substituted with strongly polarized functional) in the form of OHO interaction.37, 87 The two other two important hydrogen bonding types considered in this work are called CHπ and OHπ interaction.26, 34, 87 In particular, we investigate the trends in above interaction classes that arise, when the size and curvature of the model CNT is altered.
3.3.1 ππ interactionBetween lignin and MWCNT, ππ interaction would arise from aromatic moieties present in the lignin backbone. In our models, exclusive ππ interaction occurs exclusively when benzene adsorbs in a parallel manner on the p-SWCNT.
The very term “ππ interaction” has been scrutinized thoroughly in the literature and was subjected to controversy: Martinez and Iverson write that term like “[…] ‘π -stacking’ and ‘π–π interactions’ do not accurately describe the forces that drive association between aromatic molecules[…]”.85 Grimme on the other hand finds that “[…] π–π interactions [are] a special type of electron correlation (dispersion) effect that can only act in large unsaturated systems […].”79 Since the SWCNT used in our modeling are virtually infinite in one direction, this definition applies to the interactions that arise in our simulation.
The influence of the SWCNT curvature on ππ interaction can be seen in Figure 7.  of benzene on p-SWCNT becomes more negative—that is, binding—with increasing p-SWCNT size. This increase is mainly of geometrical origin as decreasing curvature increases the effective interacting surface are with the flat benzene molecule.76 Accordingly, Figure S6 reveals, that
of benzene on p-SWCNT becomes more negative—that is, binding—with increasing p-SWCNT size. This increase is mainly of geometrical origin as decreasing curvature increases the effective interacting surface are with the flat benzene molecule.76 Accordingly, Figure S6 reveals, that  of benzene adsorbing in a parallel manner is linearly correlated (R2 ~ 1) to the respective p-SWCNT's curvature. A theoretical maximum interaction would be achieved with graphene, which has a curvature “approaching zero.” The dispersive component and the overall ππ interaction grow with decreasing curvature (i.e., growing CNT size/diameter) while the polar component diminishes (cf. Table S3). The same applies to the case of the sw-SWCNT-adsorption of benzene (R2 ~ 0.99; cf. Figure 7). The trend observed is in agreement with prior literature.45, 76, 88-90 There is a numerical deviation at higher curvature, that is, smaller radii, where we observe significantly stronger binding than reported by Tournous and Charlier45 Their results were obtained by means of a partially correlation-corrected LDA,91 which is known for minor parametrization issues and an artificial discontinuity of second order (and higher) correlation energy correction terms at low densities.92 The latter could potentially “cut off” noncovalent interaction effects at certain distances from the C-atoms and would be noticed especially at higher curvatures.
of benzene adsorbing in a parallel manner is linearly correlated (R2 ~ 1) to the respective p-SWCNT's curvature. A theoretical maximum interaction would be achieved with graphene, which has a curvature “approaching zero.” The dispersive component and the overall ππ interaction grow with decreasing curvature (i.e., growing CNT size/diameter) while the polar component diminishes (cf. Table S3). The same applies to the case of the sw-SWCNT-adsorption of benzene (R2 ~ 0.99; cf. Figure 7). The trend observed is in agreement with prior literature.45, 76, 88-90 There is a numerical deviation at higher curvature, that is, smaller radii, where we observe significantly stronger binding than reported by Tournous and Charlier45 Their results were obtained by means of a partially correlation-corrected LDA,91 which is known for minor parametrization issues and an artificial discontinuity of second order (and higher) correlation energy correction terms at low densities.92 The latter could potentially “cut off” noncovalent interaction effects at certain distances from the C-atoms and would be noticed especially at higher curvatures.
 The
The  of benzene adsorbing on SWCNT in parallel mode is plotted—As circles—Against the nanotubes' curvature. Reference energies for either graphene, a graphene nanoribbon or armchair p-SWCNT. Are plotted as diamonds. The inset depicts the adsorption of benzene onto a (12,12) p-SWCNT
3.3.2 Comparison of CHπ hydrogen bonding and ππ interaction in simple aliphatic and aromatic lignin model molecules
of benzene adsorbing on SWCNT in parallel mode is plotted—As circles—Against the nanotubes' curvature. Reference energies for either graphene, a graphene nanoribbon or armchair p-SWCNT. Are plotted as diamonds. The inset depicts the adsorption of benzene onto a (12,12) p-SWCNT
3.3.2 Comparison of CHπ hydrogen bonding and ππ interaction in simple aliphatic and aromatic lignin model molecules
Between lignin and MWCNT, CHπ hydrogen bonding would arise between the various hydrogen atoms present in the CH3-units of lignin backbone interacting with the aromatic surface of the CNT. In our models, this situation can be mimicked through the α- and δ-adsorption of the aliphatic rings on p-SWCNT. In Figure 8, the resulting  are plotted in dependency of the p-SWCNT's curvature.
are plotted in dependency of the p-SWCNT's curvature.

Comparison of CHπ hydrogen bonding (graphs 1–6) and ππ interaction (graph 7) in simple aliphatic and aromatic lignin model molecules. The graphs names are composed of a prefix indicating the adsorbing position (s.a.) and the name of the molecule adsorbing onto the p-SWCNT (all connecting lines are just given as visual guidance). The encountered Eads are larger for p-mode adsorption
The magnitude of CHπ interaction experienced by parallel-adsorbing cyclohexane (graph 6 in Figure 8) is similar to ππ interaction experienced by parallel-adsorbing benzene (graph 7 in Figure 8). If the CHπ interaction of p-adsorbing cyclohexane is normalized to the number of adsorbing CH3-units, it can be approximately classified as “weak” hydrogen bonding (i.e., below 4 kcal/mol resp. 170 meV).93 If the adsorbing molecules adsorb from α- or δ-position (graphs 1–5 in Figure 8), the CHπ interaction energies cross the border to “medium” hydrogen bond (i.e., between 4 and 15 kcal/mol resp. 170–650 meV).93
The CHπ interaction in all models decreases with increasing curvature of the p-SWCNT. This is generally due to a simultaneous decrease in attractive dispersive interaction and increase in repulsive polar interaction. Thus, the CHπ interaction exhibits the same growth-dependence as the ππ interaction (see above).
Comments (0)