
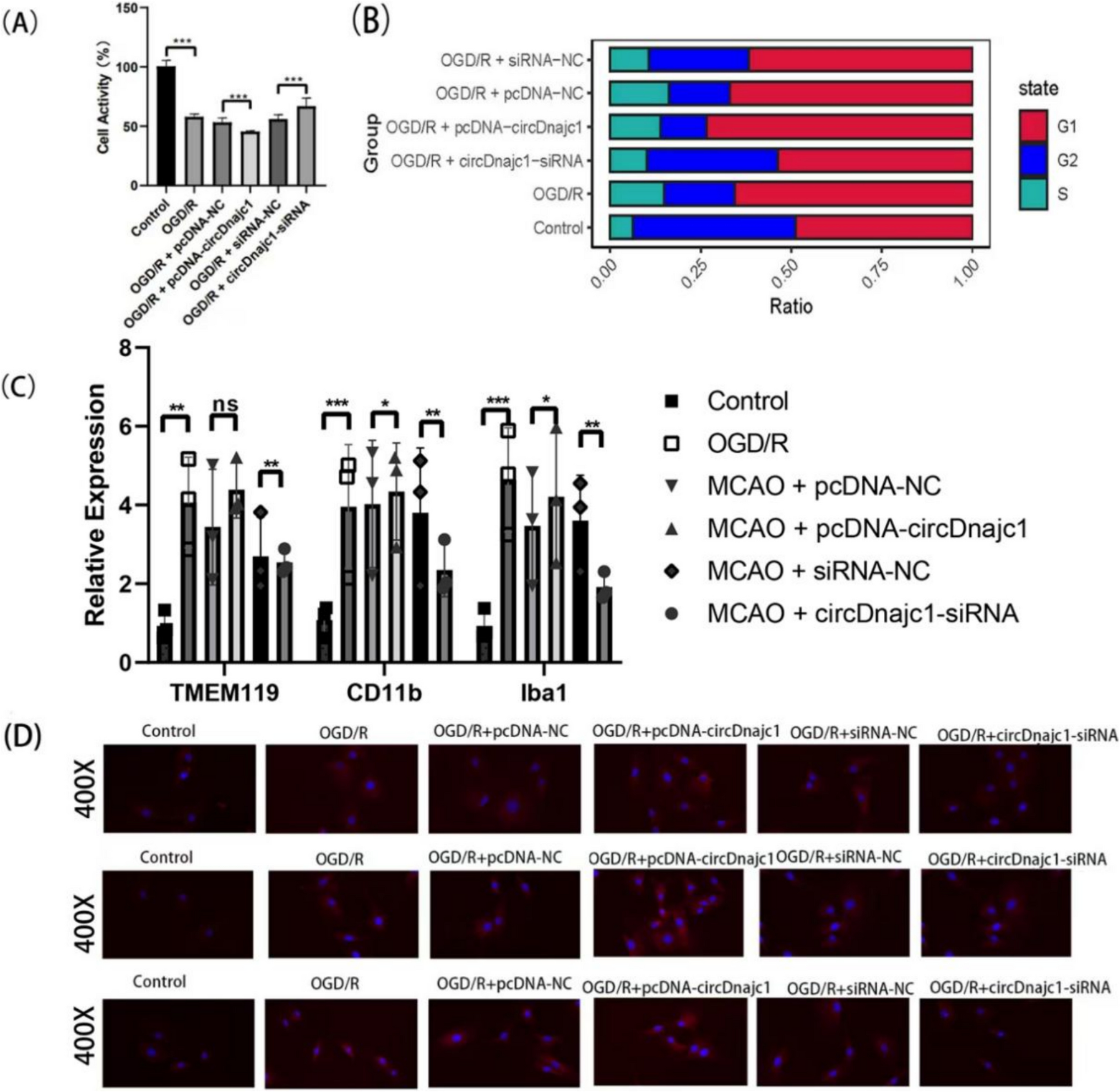
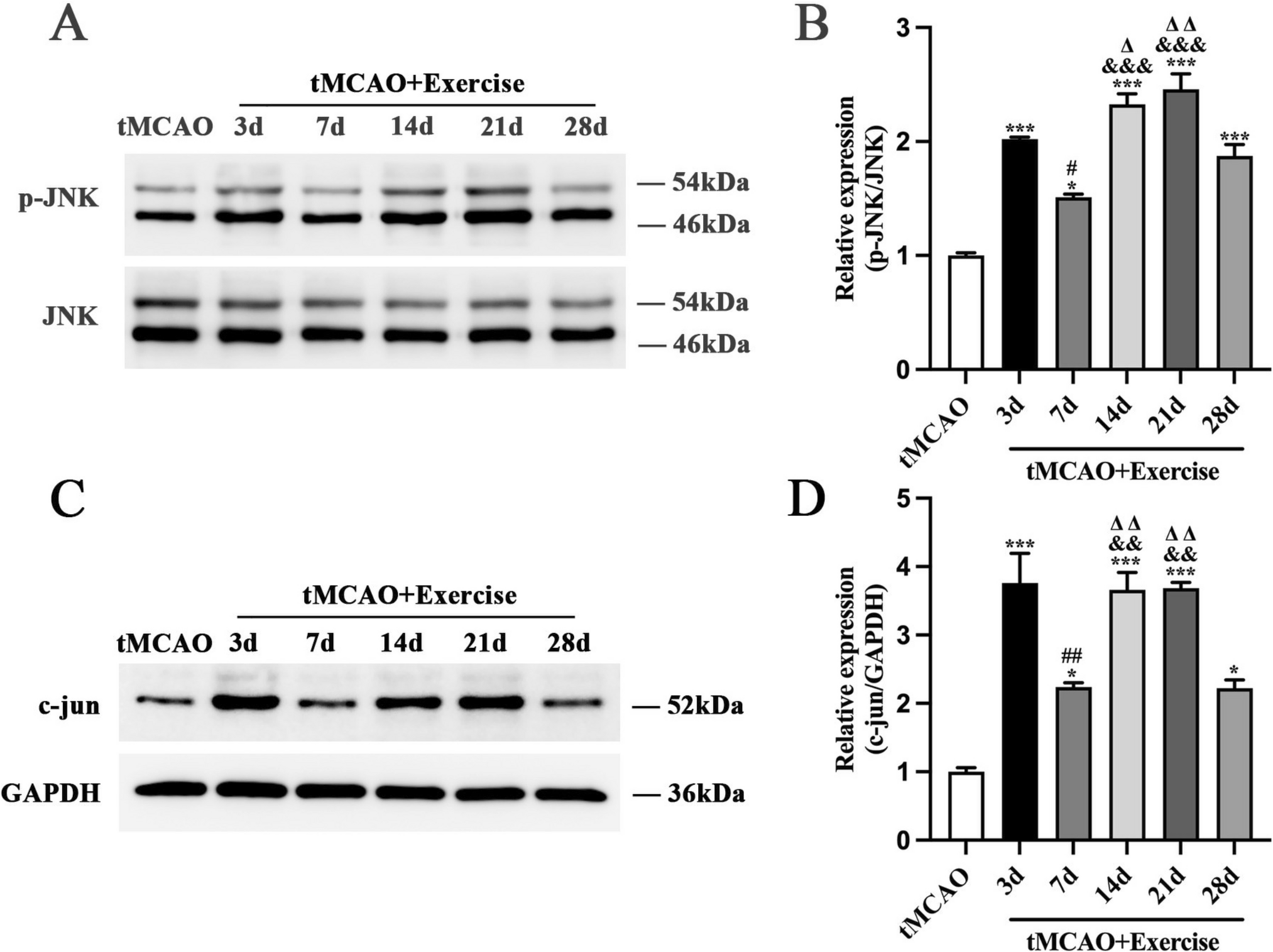
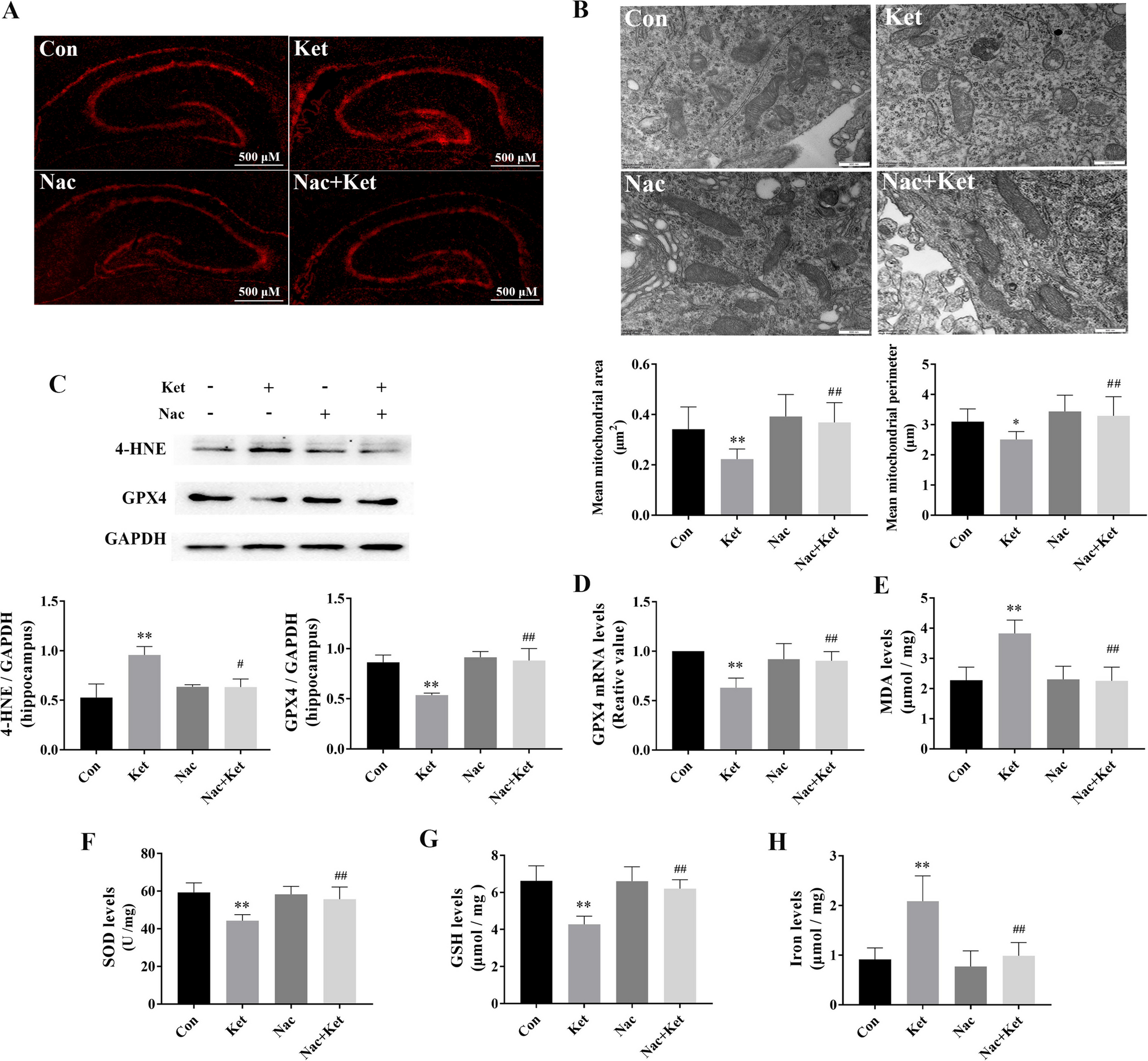
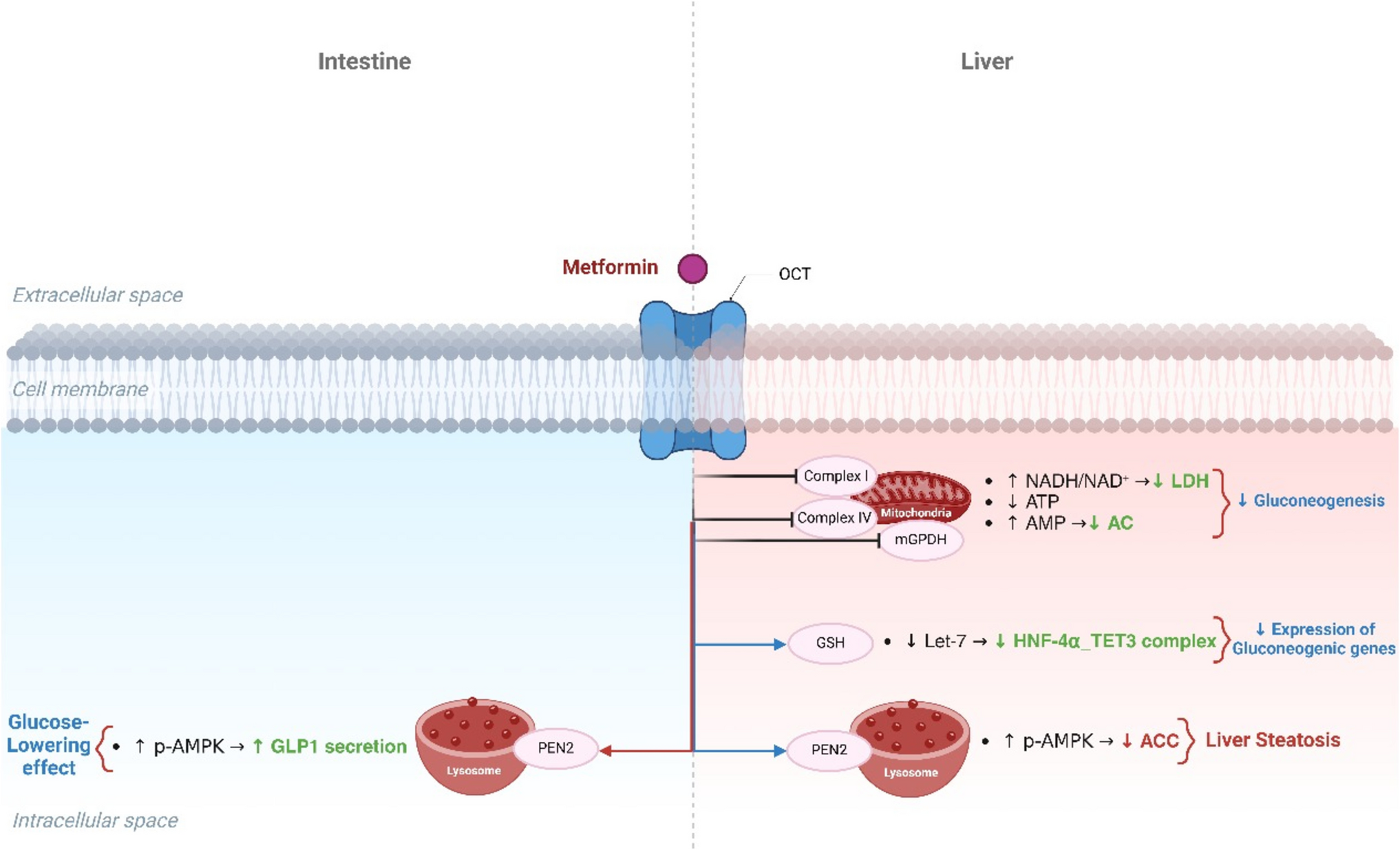
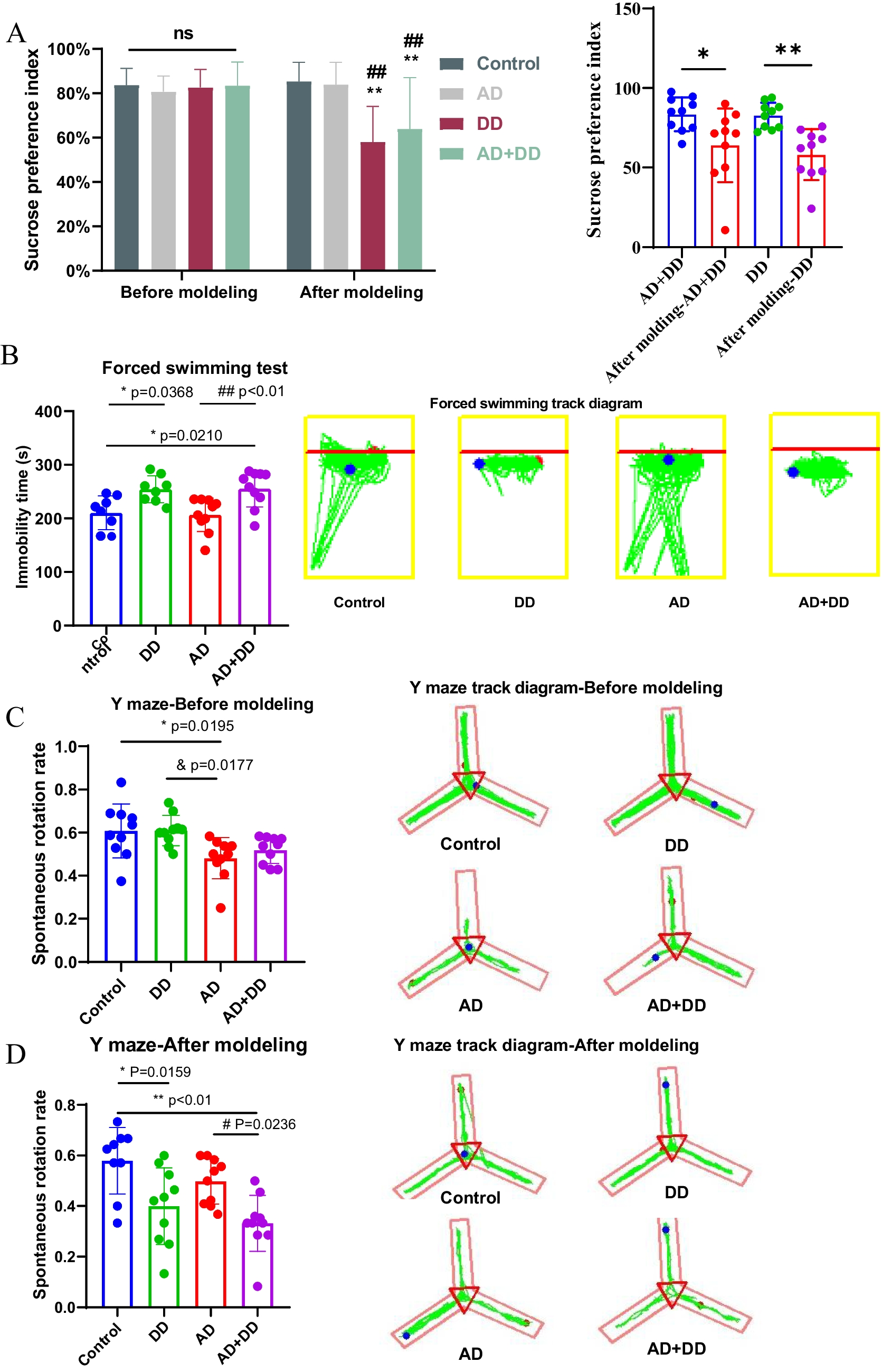
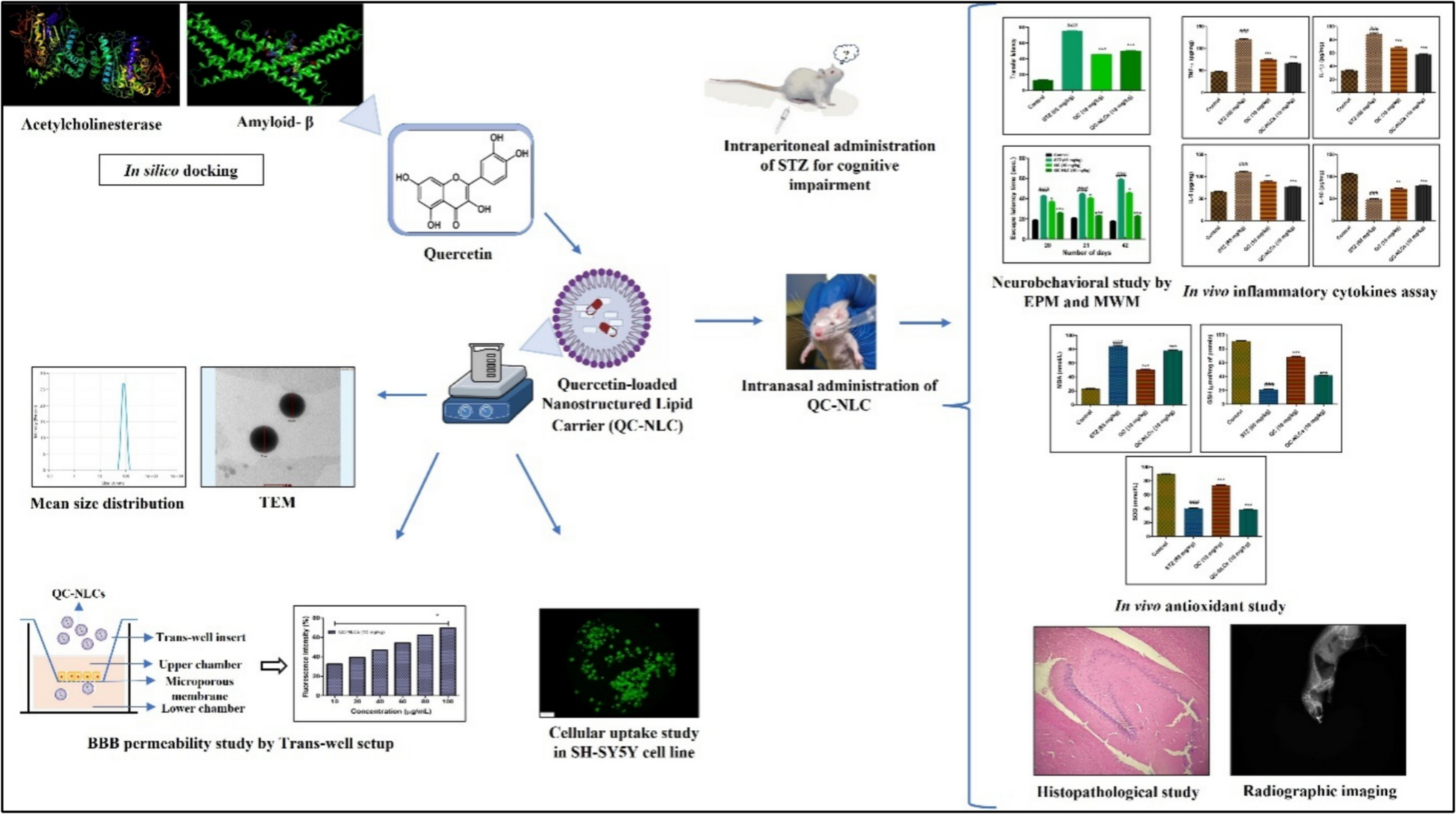
Remember me




All experiments were performed with a protocol authorized by the 2nd Local Ethical Committee for Animal Experiments in Warsaw (authorization no. WAW2/081/2018) in accordance with the EU Directive 2010/63/EU. All experiments and methods were carried out according to relevant regulations and ARRIVE guidelines.
Neonatal hypoxia–ischemia was induced in Wistar rats of both sexes on postnatal day 7 (P7) by permanent unilateral ligation of the common carotid artery, followed by systemic hypoxia according to the method proposed by Rice et al. [19]. In our previous studies, it was explored if there is a sexually dimorphic response to HI treatment [20]. As we did not notice any significant differences between male and female rats in the previous experiments, pups of either sex were used. Rats from each litter size 10–12 were randomly selected into groups (control or HI). Then, the pups were anaesthetized with isoflurane (4% induction, 2.0% maintenance), and after that, the left common carotid artery was exposed and cut between double ligatures of silk sutures. The incision was sutured and treated with lignocaine. Sham-operated (control) animals will undergo the same surgical procedure without the ligation of the carotid artery. The anesthesia time was no longer than 5 min, to avoid the neuroprotective influence of isoflurane. Then, the rat pups were returned to the mother in the home cage nest for 1 h. After that hypoxia was induced by placing the animals in a chamber at a controlled temperature (35 °C) and subjecting them to a mixture of 7.5% oxygen in nitrogen for 1 h. When the entire procedure was ended, the rats were returned to their home cage housed at 22 ± 2 °C and 55 ± 5% humidity following a 12/12-h light/dark cycle with constant access to food. In this model, hypoxic-ischemic occlusion was observed in the cerebral hemisphere on the side of the ligated artery (ipsilateral), and only these hemispheres were taken to the next experiments. The undamaged hypoxic hemisphere, as well as sham-operated animals, served as controls. The animals were randomly divided into 4 experimental groups according to the following pattern (3–5 rats per group and time point): (1) control animal (non-hypoxic, non-ischemic); (2) control animals treated with sodium butyrate, to assess the incidence of unintended deleterious effects of SB administration in the control groups (Ctr + SB); (3) post-HI animals (HI); (4) post-HI animals treated with sodium butyrate (HI + SB) (Fig. 1). During the experiment, animal mortality did not occur outside of planned endpoint. The brains for further procedures were isolated at 1, 3, 5, 7, and 14 days after HI. For qPCR and Western blot analysis, 64 rats were used (4 groups and 4 time points 1, 3, 5 and 14 days after surgery); for TEM analysis 20 rats were used (4 groups and 1 time point 7 days after surgery): for the immunohistochemical studies 20 rats were used (4 groups and 1 time point 14 days after surgery).
Fig. 1
Diagram of the in vivo model. Created with BioRender.com
Pharmacological StimulationSodium butyrate (Sigma-Aldrich) was administered subcutaneously at a dose of 300 mg/kg/day for 5 consecutive days (starting immediately after HI induction) to both control animals (Ctr + SB) and post-HI animals (HI + SB). The optimal doses and timing of sodium butyrate administration were determined in our previous experiments [20].
Tissue PreparationAt selected time points after HI or controls in match age (7 and 14 days), rats were deeply anaesthetized with intraperitoneal injections of ketamine (100 mg/kg b.w.) combined with xylazine (10 mg/kg b.w.).
For immunohistochemical studies, rats were perfused through the heart with PBS buffer (pH 7.4) followed by a fixative solution of 4% paraformaldehyde in PBS. Then, brains were isolated, postfixed for 3 h at 4 °C in the same solution, cryoprotected in 30% solution of sucrose overnight, and afterward, frozen on dry ice. The hemispheres were then cut (from Bregma − 2.80 mm Lambda 4.80 mm to Bregma 3.40 mm Lambda 4.20 mm) in regions of the cortex and hippocampus into cryostatic slices 30 mm thick, to create 10 serial sections. The brain sections were placed in a 24-well plate with a non-freezing solution (Sigma-Aldrich) and stored at − 20 °C until use.
Molecular (qPCR) and biochemical (Western blot) analyses were performed on non-perfused tissue 1, 3, 5, and 14 days after HI induction or controls in matched age. The animals were anesthetized as described above and decapitated. The hemispheres were dissected, immediately frozen on dry ice, and stored at – 80 °C until use. Only the ipsilateral (hypoxic-ischemic) hemispheres (from HI animals) and the left hemispheres (from control animals) were analyzed.
Transmission Electron MicroscopyFor transmission electron microscopy, the animals were perfused through the heart with PBS buffer (pH 7.4) followed by a mixture of 2.5% glutaraldehyde (GA), 2% PFA, and 0.1 M cacodylate buffer, and then, the brains were isolated and incubated at 4 °C for 24 h in the same solution. After a 24-h incubation, the frontal cerebral cortex (the brain region most affected by HI) was isolated from the brains and cut into small fragments. The tissue then was washed three times in 0.1 M cacodylate buffer and post-fixed in 1% osmium tetroxide (OsO4) and 0.8% potassium ferricyanide (K4(FeCN)6) for 2 h. The tissue was dehydrated in a series of alcohols of increasing concentration (30–99.8%) and in propylene oxide. The samples were then embedded in resin blocks and polymerized at 60 °C for 24 h. The polymerized material was cut into ultra-thin Sects. (40–60 nm) on an MTXL ultramicrotome, using a diamond knife, placed on copper grids (300 mesh) and post-stained using the double contrast method with uranyl acetate (30 min) and lead citrate (15 min). Analysis was performed using a JEM-1011 transmission electron microscope (JEOL) operating at an accelerating voltage of 80 kV. The level of damage to the cytoarchitecture of the cerebral cortex was evaluated, with particular attention to synaptic connections. Transmission electron microscopy studies were performed in cooperation with the Electron Microscopy Research Unit, MMRI PAS.
Western Blot AnalysisExpression of complement and pre- and postsynaptic proteins were determined by Western blot analysis in brain hemispheres after HI and sodium butyrate treatment. For Western blot analysis, the brain hemispheres were homogenized in a RIPA lysis buffer (10 mM Tris–HCl pH 7.5 containing 150 mM NaCl, 1% Nonidet P40, 0.1% SDS, 1% Triton X-100, PMSF 0.1 mg/mL) supplemented with proteinase and a phosphatase inhibitor cocktail (1:100, Sigma Aldrich). The lysates were centrifuged at 13,000 × g for 10 min at 4 °C and the supernatants were collected. Total protein concentrations were assessed using a Bio-Rad DCTM protein assay kit (Bio-Rad). Samples containing 50 μg of protein were separated by SDS–PAGE and then transferred onto nitrocellulose (Amersham™ Protran™ Supported 0.45 μm NC). Each sample was tested at minimum in duplicate and each variant in three biological replicates. After blocking in 5% non-fat milk, the membranes were incubated at 4 °C overnight with primary antibodies. The primary antibodies used for the Western blot analysis: mouse monoclonal anti-PSD-95 (ThermoFisher, 1:2000), mouse monoclonal anti-synaptophysin (Sigma, 1:1000), and rabbit polyclonal anti-synapsin I (ThermoFisher, 1:1000). In the next step of Western blot analysis, the membranes were rinsed 3 times in TBST buffer (1 × Tris-Buffered Saline, 0.1% Tween® 20 Detergent) and then incubated for 1 h at room temperature with an anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich). To verify an equal protein loading per line, a mouse monoclonal anti-β-actin (UniProtKB, 1:1000) was used as an internal control for each Western blot analysis. Immunoblot signals were visualized using an ECL chemiluminescence kit (GE Healthcare Life Sciences) by exposure of the membrane to an X-ray HyperfilmTM ECL film (GE Healthcare Life Sciences). A semiquantitative estimation of protein levels detected by immunoblotting was performed utilizing LKB Utrascan XL Program GelScan software. The densitometry values were averaged in all groups and then the densitometry values in the control groups were taken as 100%. The data from the respective experimental groups are presented as percentages of the control value.
Immunohistochemical EvaluationIn order to determine the effect of sodium butyrate on the modification of synaptic networks and expression/localization of selected complement proteins in brains after HI injury, double immunohistochemical staining was performed using antibodies labeling selected complement proteins C3 (ThermoFisher/Invitrogen) and C5 (ThermoFisher/Invitrogen) as well as proteins localized in postsynaptic membrane PSD-95 (ThermoFisher/Invitrogen).
The brain sections were placed in a new 24-well plate and washed with PBS. Subsequently, the slices were incubated in a blocking medium (10% normal goat serum in PBS containing 0.1% Triton X-100) for 1 h at room temperature. The sections were incubated overnight at 4 °C with a primary antibody specific for complement proteins (goat polyclonal anti-C3 (1:500) or rabbit polyclonal anti-C5 (1:100)). Subsequently, the slices were rinsed in PBS, exposed to the appropriate Cy3-conjugated secondary antibodies (AlexaFluor 546, 1:500) for 1 h at room temperature in the dark. In the next step of double immunofluorescent staining, sections were washed 3 × 5 min in PBS and incubated with primary antibody for synaptic proteins (mouse monoclonal anti-PSD-95 (1:500)) overnight at 4 °C. Then, after rinsing in PBS, the primary antibodies were revealed by applying the appropriate secondary FITC-conjugated antibodies (AlexaFluor 488, 1:500) for 1 h at room temperature. After washing the slices in PBS (3 × 5 min.), the nuclei were labeled with the fluorescent dye Hoechst 33258 (2 μg/ml PBS; Sigma–Aldrich) for 20 min and washed in PBS. Finally, sections were removed from the wells, applied to microscope slides, coated with Fluorescent Mounting Medium (Dako), and microscope cover glasses, and then dried. Labeling was verified using a confocal laser scanning microscope (LSM 780, Carl Zeiss, Germany) with ZEN software. A helium–neon laser (543 nm) was utilized in the excitation of Alexa Fluor 546, while an argon laser (488 nm) was applied in the excitation of FITC. Images were collected from three different fields of view.
Reverse Transcription and Quantitative PCR AnalysisThe effect of sodium butyrate on gene expression of selected complement proteins (C1q, C3, C5, C9) and their receptors (C3aR, C5aR) was determined in brains of HI-treated and control animals in different experimental time points (24 h, 72 h, and 5 days after surgery procedure).
Total RNA was isolated using the Total RNA Mini Kit (A&A Biotechnology), according to the manufacturer’s instructions. The quality and concentration of RNA were verified by spectrophotometry with the Nanodrop™ apparatus. The samples containing 1 μg of total RNA were reverse transcripted using high-capacity RNA-to-cDNA Kit (Applied Biosystems) according to the manufacturer’s recommendation. The analysis of changes in the mRNA level of genes was carried out using FAST SYBR Green Master Mix Reagent (Applied Biosystems), 2 μl of cDNA samples, and specifically designed primers (Table 1). The quantitative PCR reaction was performed in the 7500 Fast real-time PCR System (Applied Biosystems). The reaction steps were as follows: (1) holding stage, 10 s at 95 °C; (2) cycling stage (40 ×), 3 s at 95 °C, 30 s at 60 °C, and 45 s at 72 °C; and (3) melt curve stage, 15 s at 95 °C, 1 min at 60 °C, repeated in two cycles. Each sample was tested in triplicate, and each variant was in three biological replicates. The dissociation curve will be plotted to determine the specificity of the amplification. The fluorescence signals of a specific transcript were normalized against those of the reference gene (β-actin), and the threshold cycle values (ΔCt) were quantified as fold changes using the 2−ΔΔCT method.
Table 1 List of designed primers used in quantitative real-time PCR analysisStatistical AnalysisExperimental groups were coded (named by numbers) for the person who made the statistical analysis to reduce bias. Data analysis was performed using dedicated statistical software (GraphPad Prism 8.0). All results were presented as mean values from individual experimental data, ± standard deviation (SD). Statistical significance analyses were performed on data obtained from at least 3 experiments and 3 technical replicates. Comparisons between groups were performed using two-way analysis of variance (ANOVA) followed by the Bonferroni post-hoc test for multiple comparisons. The data were considered statistically significant at p-value < 0.05. Power analysis: The sample sizes for each of the studied groups were determined with the assumptions: 99,5% confidence level, 90% power of the test. With these parameters, the minimum required sample size in each group was 3. The numbers were determined based on preliminary data, assuming that for the complement protein C5 index, which is one of the key proteins in the study, the difference between means will be 30 and the standard deviation will be SD = 12.
Comments (0)