Mice and ethics statement
Six- to eight-week-old BALB/c female mice were procured from SPF Biotechnology Co., Ltd. (Beijing, China) and bred under specific pathogen-free conditions following standard and approved protocols. All animal procedures were conducted in compliance with the Ethics Review Committee of the National Institute for Communicable Disease Control and Prevention at the Chinese Center for Disease Control and Prevention (protocol code 2020–018). All efforts were made to minimize suffering.
Bacterial strains and culture conditions
N. farcinica IFM10152 strain was procured from the German Resource Centre for Biological Materials. Twenty-three N. farcinica isolates were randomly selected from our laboratory and cultured in BHI broth (Oxoid Ltd, UK) at 37 °C. Escherichia coli BL21(DE3) was procured from TransGen Biotech and cultured in LB medium containing kanamycin (50 µg/ml). All strains were cultured to exponential phase before experiments.
Conservative and bioinformatic analysis
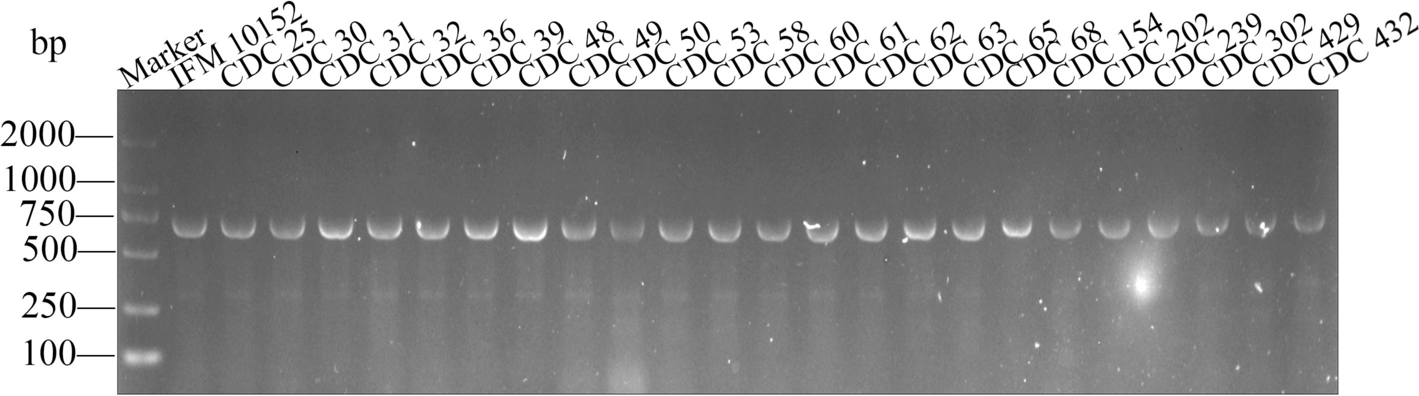
For conservative analysis, the nfa47630 sequence was matched with N. farcinica sequences by NCBI Nucleotide Blast. To further verify its conservation in clinical strains, DNA was extracted from N. farcinica IFM10152 and 23 isolated N. farcinica strains (Table S1) using the Wizard Genomic DNA Purification Kit (Promega, USA), and PCR was performed using nfa47630 specific primers: forward 5'-ATCTGAATTCATGCCGACCGCAGTAG-3' and reverse 5'-ATGTAAGCTTTCAGGCGACGGTGAAG-3'. PCR products were then detected by 2% agarose gel electrophoresis. The bioinformatic analysis of NFA47630 was performed by searching (I) subcellular localization using PSORTb (https://www.psort.org/psortb/) [19], (II) signal peptide using UniProt (https://www.uniprot.org/) [20], (III) transmembrane helices using HMMTOP (http://www.enzim.hu/hmmtop/) [21], and (IV) antigenic propensity using Protein Variability Server (http://imed.med.ucm.es/PVS/) [22].
Expression and purification of recombinant NFA47630 proteins
For preparation of recombinant proteins, plasmids were cloned into the pET-30a( +) expression vector by digestion with EcoR I and Hind III and transformed into Escherichia coli BL21(DE3). Recombinant E. coli BL21 cells were selected and grown in LB medium containing 50 µg/ml kanamycin at 37 °C for 6 h until the OD600 reached 0.8. Then, protein expression was induced by the addition of 0.2, 0.5 and 1 mM IPTG to the bacterial culture for 4 h at 28 and 37 °C. After the bacterial suspension was sonicated with protease inhibitor, the pellet and culture supernatant were obtained by centrifugation at 12,000 rpm/min for 20 min at 4 °C, and all protein preparations were analyzed by SDS‒PAGE and quick blue staining. The recombinant NFA47630 (rNFA47630) protein was then purified using the His-Bind purification kit (Novagen, Germany). After the collected protein was eluted by a Ni–NTA column with elution buffer, nonspecific proteins were removed by washing with 20, 40, 60, 100, and 250 mM imidazole buffer. All eluted fractions were collected and analyzed by SDS–PAGE and quick blue staining. Purified protein was then concentrated with a 10-kDa centrifugal filter device (Millipore, MA), and endotoxin was further removed using a ToxinEraser endotoxin removal kit (GenScript, China) according to the manufacturer’s guidelines. The protein was stored at -80 °C after concentration determination by a bicinchoninic acid (BCA) assay.
Antigenicity determination by western blot
Total rNFA47630 proteins were separated by SDS–PAGE (5–12%) and transferred onto 0.45 µm polyvinylidene fluoride (PVDF; Merck, Germany) membranes. The membranes were then blocked with 5% skim milk diluted in TBS containing 0.05% Tween (TBST) for 2 h at room temperature. Mouse antisera (stored in our laboratory) included N. farcinica, N. asteroids, N. cyriaciegeorgoca, N. brasiliensis, or Mycobacterium bovis, and horseradish peroxidase (HRP)-conjugated monoclonal anti-pentahistidine (His) antibody (New England Biolabs Inc., USA) was diluted (1:2,000) in TBST containing 2% skim milk and incubated overnight at 4 °C. The membranes were then washed with TBST three times and incubated in an HRP-conjugated goat anti-mouse IgG (1:4000; Southern Biotech, USA) secondary antibody for 1 h at room temperature. After three washes with TBST, the bands were visualized using Amersham® Hyperfilm® ECL™ and MP Autoradiography Films (GE Healthcare).
Cell culture
The mouse macrophage cell line RAW264.7 was procured from the National Infrastructure of Cell Line Resource (Beijing, China) and cultured in high-glucose DMEM (Gibco, NY, USA) supplemented with 10% FBS at 37 °C. In each experiment, cells from passages 5 to 15 were counted before seeding in the plate.
MAPK signaling pathway analysis
For MAPK signaling pathway analysis, RAW264.7 cells were seeded in 6-well microplates at a density of 8 × 105 cells per well for 16–18 h and then stimulated with 2, 4 or 8 μg/mL rNFA47630 protein. In order to exclude residual LPS interference, rNFA47630 preparation was pretreated with 100ug/ml polymyxin B (PmB, a specific inhibitor for LPS, INALCO, USA) at 37 °C for 2 h [23]. Additionally, 100 ng/mL LPS (with or without 100 μg/mL PmB) was added to the cell plate as positive control. At indicated time points, whole-cell extracts were harvested using RIPA lysis buffer (strong) (CWBIO, Beijing, China) containing protease and phosphatase inhibitor cocktail for Western blot analysis. Equal amounts of protein were separated by SDS–PAGE and transferred to PVDF membranes as described before. Primary antibodies against p-ERK1/2 (1:1000, CST, USA), p-JNK (1:1000, CST, USA), p-P38 (1:1000, CST, USA), and β-actin (1:4000, CST, USA) were diluted in TBST containing 2% skim milk. HRP-conjugated goat anti-rabbit IgG (1:1000, Beyotime, China) or HRP-conjugated goat anti-mouse IgG (1:4000, ZSGB-BIO, China) was used as the secondary antibody.
Cytokine measurements
To measure cytokines in rNFA47630-stimulated RAW264.7 cells, cells were seeded in 24-well culture microplates at a density of 2 × 105/well and 2 μg of rNFA47630 was added for 6, 18 and 24 h. Similarly, we set up rNFA47630 + PmB and LPS (with or without PmB) groups to test residual LPS effects. To block MAPK signaling, cells were pretreated for 1 h with inhibitors of 20 µM ERK (PD 98059, Sigma, USA), 20 µM JNK (SP 600125, Sigma, USA) or 20 µM p38 (SB 203580, Sigma, USA) prior to rNFA47630 protein exposure. Then, culture supernatants were harvested at the indicated times, and the cytokine concentrations were determined by TNF-α, IL-10, IL-12 and IFNγ ELISA kits (BD OptEIA™, USA) according to the manufacturer’s instructions.
Mouse immunization
Female mice were randomly divided into rNFA47630 and PBS groups and immunized by intramuscular injection of 100 µl recombinant protein-aluminum hydroxide adjuvant mixture (10 µg of rNFA47630 per mouse) or equivalent PBS-aluminum hydroxide adjuvant mixture three times every 2 weeks. Whole blood (with the addition of the anticoagulant heparin) and sera in rNFA47630-immunized (n = 6) and PBS-immunized mice (n = 6) were collected 7 days after the last immunization, and rNFA47630-specific IgG, IgG1, IgG2a and IgG2b (Abcam, UK) antibodies were determined by ELISA. To confirm antibody production during infection, rNFA47630 protein was separated by SDS–PAGE and transferred to PVDF membranes as described before. Anti-rNFA47630 sera (1:2000) were used as the primary antibodies, followed by a secondary antibody (HRP-conjugated goat anti-mouse IgG).
Whole blood and neutrophil killing assay
Equal whole blood (with the addition of the anticoagulant heparin) from rNFA47630- and PBS-immunized mice was collected and immediately mixed with a 1 × 106 CFU N. farcinica suspension in a 37 °C incubator for 2 h. Then, serial dilutions of mixtures were plated on BHI agar plates for CFU count.
Mice were then sacrificed by cervical dislocation, and femurs and tibias in PBS-immunized mice were dissected and flushed out with HBSS (Solarbio, China) for bone marrow cell isolation. Subsequently, marrow suspensions were centrifuged at 500 × g and 4 °C for 10 min, and the pellets were resuspended with 45% Percoll and overlaid onto Percoll gradient layers (81%, 62%, 55%, and 50%). After centrifugation at 500 × g for 30 min, cells were collected from the 81%/62% interface, and red blood cells were depleted using RBC lysis buffer (BD OptEIA™, CA, USA). Bone marrow neutrophils were then counted and incubated in a 24-well microplate at a density of 2 × 105 cells per well with RMPI 1640 medium supplemented with 10% FBS. After 24 h of stimulation with 2 μg of rNFA47630, N. farcinica suspension was added to each well at a ratio of 10:1 for 2 h of incubation, and serial dilutions of whole cell lysates were then plated on BHI agar plates for bacterial survival determination.
Spleen cell isolation
Spleens in PBS-immunized mice were dissected and transferred into a 40 µm cell strainer placed within a 50 ml tube, then mashed gently with a 5 ml syringe plunger and washed repeatedly with 1 × HBSS. After centrifugation at 500 × g for 10 min twice and depleting red blood cells using RBC lysis buffer, spleen cells were incubated in a 24-well microplate with RMPI 1640 medium supplemented with 10% FBS and stimulated with 2 μg of rNFA47630. Supernatants were collected after 1, 2, and 3 days of incubation, and the cytokine concentrations of IFN-γ and IL-4 were determined by ELISA kits as described above.
Mouse challenge
Ten days after the last immunization, mice in the rNFA47630 (n = 10) and PBS (n = 10) groups received 50 µl of N. farcinica (1 × 107 CFU) suspension through intranasal inoculation. Loss of weight and elevated body temperature were quantified immediately prior to infection and 24 h postinfection as previously described. Pulmonary bronchoalveolar lavage fluid (BALF) was obtained through 3 successive lavages of the bronchi with ice-cold PBS for lactate dehydrogenase (LDH) assessment using the LDH-Glo™ Cytotoxicity Assay (Promega, USA) procedures. Whole lungs were collected and homogenized and then plated on BHI agar plates by serial dilutions. Bacterial counts were enumerate after 48 h in a 37 °C incubator. Lung homogenate was then centrifuged to obtain the supernatant samples, and the concentrations of TNF-α, IL-10, IL-12, and IFN-γ were determined by ELISA as described above.
For survival rate analysis, each mice in the rNFA47630 (n = 10) and PBS (n = 10) groups were challenged by intraperitoneal injection of 100 µl (1 × 109 CFU) bacterial suspension, and mouse survival was monitored daily for a 10-day period. To assess the severity of pathological damage, surviving mice were sacrificed by cervical dislocation. Lungs, liver and brain tissues were collected and fixed in 4% paraformaldehyde, embedded in paraffin and sectioned. Subsequently, the deparaffinized tissue sections were stained with hematoxylin and eosin (H&E). Stained sections were observed using a biological microscope (Nikon, Eclipse Ci-L, Japan), and images were randomly captured under high-power fields (20 × magnification).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9.0.0. Statistical differences were analyzed using ordinary one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons. Survival rates were analyzed by Kaplan–Meier and log rank test. For all experiments, values of differences p ≤ 0.05 were considered statistically significant.




Comments (0)