
Remember me








Organic electrochemistry is gaining increasing interest in both academia and industry due to its numerous advantages and potential applications . Electrochemical methods can reduce costs and waste generation by eliminating the need for chemical oxidants or reductants, and they can be safely and easily scaled up in flow reactors for industrial applications. The potential for upscaling and applicability towards LSF makes electrosynthesis particularly appealing for the fine-chemical and pharmaceutical industry. As the electrode potential can be fine-tuned over a continuous scale, higher functional group compatibility can be achieved compared to many classical methods. In light of the general trend towards more chemoselective protocols with broader functional group compatibility, there has been a growing interest in exploring the potential of electrosynthesis for the late-stage functionalization of complex scaffolds. Additionally, the increased interest of medicinal chemists in electrochemical methods, combined with their continuous search for new LSF strategies, will further extend the applicability of newly established electrochemical methods.
LSF is a rapidly growing field that offers new opportunities in drug discovery . Transition-metal-catalyzed LSF strategies have been well-established over the past decades. More recently, with the vigorous development of photochemistry and electrochemistry, numerous innovative reports on LSF using photo-, electro-, and photoelectrochemistry have emerged. These areas have been systematically summarized, classifying them by targeted C–H bond functionalization and the newly formed bonds .
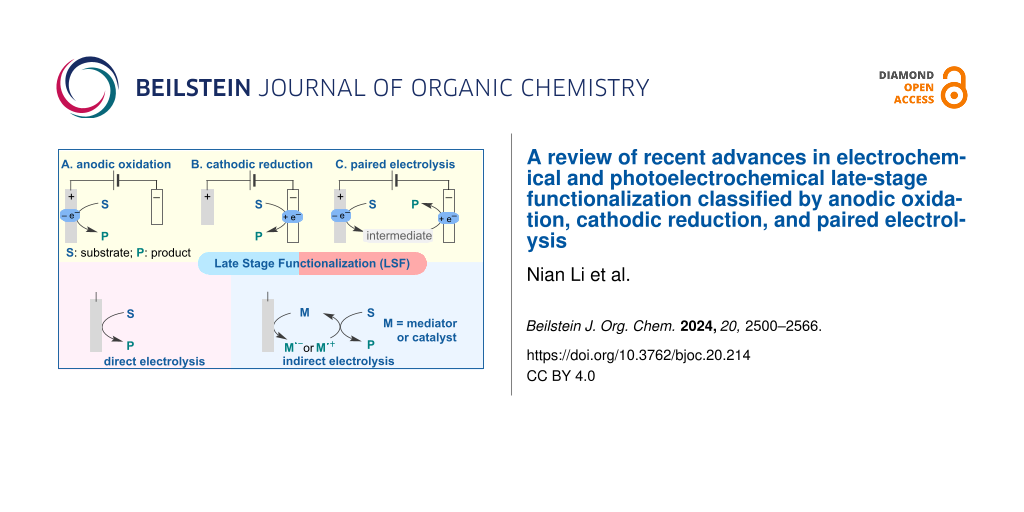
In this review, we aim to provide a comprehensive classification and overview of the currently available electrochemical and photoelectrochemical methods for the LSF of pharmaceutical drugs and natural products. We classify these advancements into three types: anodic oxidation, cathodic reduction, and paired electrolysis (Figure 1). This review considers direct electrolysis (oxidation or reduction), mediator-induced electrolysis, and metal-catalyzed and photocatalyzed electrochemical transformations. Detailed reaction conditions, such as electrolyte, electrode material, and the use of constant current or constant voltage, are presented. Additionally, we discuss the mechanisms of some representative reactions and provide selected examples of LSF of relevant bioactive compounds.
![[1860-5397-20-214-1]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-20-214-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Classification of LSF reactions in this review.
Review 1 LSF via anodic oxidationTo date, the majority of electrosynthetic methods in organic chemistry consists of anodic oxidations. These techniques are generally more robust and can often be performed outside of a glovebox, making them particularly attractive for larger scale applications in industrial settings. An anodic oxidation is frequently employed for C–H functionalization, which can simplify late-stage functionalization strategies. Additionally, many of these synthetic methods do not require precious metals, enhancing their appeal in terms of sustainability and cost-effectiveness. However, it should be noted that anodic oxidations often require electrodes with high resistance to oxidation, such as platinum electrodes, or inert electrodes with a highly developed surface, like reticulated vitreous carbon (RVC). Anodic oxidations generally involve the evolution of hydrogen (indicated in schemes as H2↑) in the cathodic half-reaction, which will however not be addressed in greater detail in this review.
1.1 Direct anodic oxidation of substrates1.1.1 C–H bond functionalization. C–H bond carbofunctionalization: CF3 groups can be installed on heteroarenes at a late stage via a TM-free electrochemical method. This route was reported in 2014 by the Baran and Blackmond groups . A commercially available reagent, Zn(SO2CF3)2, was used as the CF3 radical source in the reaction. Additionally, a series of substrates could be difluoromethylated under the reported electrochemical conditions. A comparison was made between the developed electrochemical conditions for each substrate and an analogous non-electrochemical method using peroxide for CF3 radical generation. In all cases, the electrochemical route delivered improved yields (Scheme 1).
![[1860-5397-20-214-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C(sp2)–H trifluoromethylation of heteroarenes.
The Wang group later discovered a C(sp2)–H functionalization method where primary, secondary, and tertiary alkyl radicals can be readily generated through the sequential anodic oxidative fragmentation of alkyl carbazates, enabling the functionalization of N-heteroarenes . This transformation is particularly valuable as the cleavage of the C–O bond to activate alcohols presents a significant synthetic challenge. The carbazate substrates are easily prepared from ubiquitous alcohol precursors. The first stage of the transformation involves the sequential anodic oxidation of the carbazate and subsequent deprotonation to form a diazenecarboxylate. Further anodic oxidation cleaves the diazene, resulting in the formation of an acyl radical and the release of molecular nitrogen. The subsequent step involves the decarboxylation of the acyl radical to produce an alkyl radical. This method was successfully applied to the late-stage functionalization of bioactive compounds such as caffeine and prothioconazole (Scheme 2a). Additionally, Lin, Terrett and Neurock's group reported the electrochemical C(sp3)–H methylation of complex molecules. This strategy enabled the synthesis of the "magic methyl" product, a TRPA1 antagonist (Scheme 2b).
![[1860-5397-20-214-i2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: C(sp2)–H and C(sp3)–H alkylation of complex molecules.
C–H bond amination: Direct and selective CH-aminations and amidations are challenging reactions. In this context, the regioselective sulfonamidation of (hetero)aromatic groups was achieved by the Lei group via dehydrogenative aryl C–H/N–H cross-coupling . A crucial step in this transformation is the generation of sulfamidyl radicals via a concerted proton-coupled electron transfer (PCET). This process occurs after the formation of a hydrogen bond between dibenzenesulfonimide and n-Bu4NOAc. The formed sulfamidyl radical can directly react with the (hetero)aromatic ring. Subsequent anodic oxidation produces a carbocation intermediate, which rearomatizes through proton loss. Concurrently, the cathodic reduction of the generated protons produces H2. In addition to (hetero)aromatic groups, alkene scaffolds also underwent this reaction (Scheme 3).
![[1860-5397-20-214-i3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Electrochemical oxidation-induced intermolecular aromatic C–H sulfonamidation.
In the same year, the Lei group extended the electrochemical C(sp2)–H functionalization C–N coupling reaction by developing an electrochemical method for the bioconjugation of tyrosine in proteins/polypeptides with phenothiazine residues, achieving excellent site- and chemoselectivity (Scheme 4a). This method was inspired by an earlier work from the Gouin group, which reported the merger of electrochemistry and bioconjugation in 2018 (Scheme 4b) .
![[1860-5397-20-214-i4]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Bioconjugation of tyrosine with (a) phenothiazine and (b) urazole derivatives.
In 2020, Zheng and coworkers developed an interesting iodoamination of indoles using unactivated amines and benzotriazoles . This difunctionalization reaction was carried out in an undivided cell with an RVC anode and a foamed Ni cathode, at a constant current of 12 mA in DMSO at room temperature under atmospheric conditions. The reaction has been applied to more than 80 examples, including the late-stage functionalization of natural products and pharmaceuticals, as well as the synthesis and radiosynthesis of ¹³¹I-labeled compounds. For example, the late-stage iodoamination of cytisine, amoxapine, and fluoxetine hydrochloride was achieved with yields of 65%, 87%, and 73%, respectively. Additionally, this transformation was successful for gram-scale synthesis via batch and flow chemistry, indicating significant potential for further industrial and medicinal chemistry applications (Scheme 5).
![[1860-5397-20-214-i5]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Electrochemical iodoamination of indoles using unactivated amines.
Furthermore, Ackermann and coworkers described a straightforward C(sp3)–H amination of 1,3-diarylpropenes with sulfonamides via direct oxidation of allylic C(sp3)–H bonds . During the reaction process, a radical cation is formed by oxidation of the substrate at the anode. This radical cation is subsequently deprotonated to produce an allyl radical. The allyl radical is further oxidized to form the allyl cation, which is then attacked by the nucleophilic sulfonamide, leading to the formation of the desired C–N-bond product. To demonstrate the mildness of the LSF reaction conditions, celecoxib and topiramate sulfonamides were easily functionalized with 1,3-diarylpropene in moderate yields (Scheme 6).
![[1860-5397-20-214-i6]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Allylic C(sp3)–H aminations with sulfonamides.
C–H bond oxygenation: In addition to electrochemical C–H aminations, C–H oxygenations have also been reported. For example, Liu and colleagues demonstrated the electrochemical oxidation of benzylic C–H bonds to ketones using tert-butyl hydroperoxide as the radical initiator . This method was applied to functionalize bioactive molecules, with celestolide, ibuprofen methyl ester, and papaverine being oxidized at the benzylic position in good yields. A gram-scale test was conducted to confirm the potential for large-scale applications. According to the authors, the electrochemical oxidation of t-BuOOH at the anode leads to a tert-butyl peroxyl radical that activates the C–H bond at the benzylic position of the substrate. The formed radical reacts with t-BuOOH to produce the corresponding ketone, with tert-butanol as a byproduct (Scheme 7).
![[1860-5397-20-214-i7]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Electrochemical benzylic oxidation of C–H bonds.
A closely related transformation was developed by Cheng and Xu in 2020 for the electrooxidation of methylarenes to aromatic acetals . With this method, several structurally diverse aromatic acetals have been synthesized. Dehydroabietic and norcholanoic acid derivatives have been effectively modified using the developed protocol. The reaction is reported to involve the oxidation of the benzene core, followed by electron transfer to the radical cation, and subsequent C–H abstraction. The methylarene undergoes oxidation, deprotonation, and a second oxidation before being captured by MeOH to produce a monomethoxylated product. This intermediate then undergoes a second oxidation round to yield the final product. Additionally, the same group disclosed an aromatic C–H hydroxylation process by combining continuous flow chemistry and electrochemistry (Scheme 8) .
![[1860-5397-20-214-i8]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Site-selective electrooxidation of methylarenes to aromatic acetals.
The surface modification of electrodes can lead to improved reactivity and selectivity. In this regard, Li and coworkers developed electron-deficient W2C nanocrystal-based electrodes to enhance the direct activation of C(sp3)–H bonds under mild conditions . The pronounced electron-deficient W2C nanocatalysts greatly facilitate the direct deprotonation process, ensuring the longevity of the electrode by overcoming self-oxidation. The LSF of drug molecules such as ibuprofen methyl ester and celestolide showed high yields and good selectivity (Scheme 9).
![[1860-5397-20-214-i9]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Electrochemical activation of C–H by electron-deficient W2C nanocrystals.
The Lei group also disclosed another C(sp3)–H functionalization involving C–O-bond formation . The reported method allows the straightforward preparation of α-acyloxy sulfides from ubiquitous carboxylic acids and sulfides, providing an alternative to the harsh Pummerer rearrangement. Methanol played a crucial role in achieving the desired transformation and it was suggested to promote the self-assembly of reagents 24 and 25 for the formation of 27, which allows the selective abstraction of H+ from the less sterically hindered side. Subsequently, the generated intermediate 29 is oxidized at the anode, then attacked by the acid to obtain the final product (Scheme 10).
![[1860-5397-20-214-i10]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: α-Acyloxy sulfide preparation via C–H/OH cross-dehydrogenative coupling.
C–H bond sulfur functionalization: The direct formation of the CS bond is an attractive way to prepare aryl sulfides. From this perspective, Wu and coworkers developed a method for the regioselective thiolation of aromatic C–H bonds by activating the thiol rather than the arene . For their developed reaction, Pt electrodes were used in an undivided cell with a mixture of HFIP/DCE 3:1 at room temperature under argon. Late-stage functionalization was demonstrated for atomoxetine, metaxalone, and tadalafil. Mechanistically, thiophenol is oxidized at the anode to the corresponding radical by SET, then dimerizes into a disulfide, which is further oxidized into an intermediate cation radical, yielding a highly electrophilic species. Subsequently, a selective anisole attack leads to an intermediate product, which is then deprotonated, generating the thiol radical. This allowed for the preparation of the para-thiolation product (Scheme 11).
![[1860-5397-20-214-i11]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Aromatic C–H-bond thiolation.
Sulfonamides are an important class of bioactive molecules. In 2021, the Waldvogel group disclosed the first C(sp2)–H functionalization protocol for the installation of sulfonamide groups using commercially available SO2 and amines (Scheme 12) . This method is highly appealing for industrial applications and LSF. The proposed mechanism begins with the anodic oxidation of the arene substrate. The resulting radical cation intermediate is then attacked by the nucleophilic amidosulfinate, which also functions as an electrolyte. The amidosulfinate is generated through the formation of a Lewis acid–base adduct. A subsequent oxidation step, accompanied by deprotonation, yields the sulfonamide product. SO2 captures the excess electrons via cathodic reduction and to prevent the reoxidation of the reduced SO2 at the anode, a divided cell setup is required (Scheme 12).
![[1860-5397-20-214-i12]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: C(sp2)–H functionalization for the installation of sulfonamide groups.
C–H bond halogenation: Aryl and alkyl halides are important synthetic building blocks for cross-coupling reactions as well as bioactive molecules with applications in agrochemical and pharmaceutical chemistry. In 2019, Jiao and colleagues reported that 1,2-dichloroethane (DCE) could be used as a chlorination reagent for the production of (hetero)aryl chlorides and vinyl chlorides . The reactions were carried out in an undivided cell containing a mixture of DCE in methanol, equipped with a graphite anode and a platinum plate cathode, under a current of 10 mA at 60 °C for 3–20 hours. Several electrochemical LSF of pharmacologically active molecules were tested, including naproxen methyl ester, a derivative of aminoglutethimide, and paracetamol. The corresponding products were obtained in good yields (51–81%). The reaction involves the catalytic dehydrochlorination of DCE at the cathode, simultaneously with anodic oxidative aromatic chlorination using cathodically released HCl as the chloride source (Scheme 13).
![[1860-5397-20-214-i13]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Preparation of (hetero)aryl chlorides and vinyl chloride with 1,2-dichloroethane. aCu(OAc)2 (0.05 equiv) is added.
Additionally, the Lei group demonstrated a double oxidation strategy to obtain α-chlorosulfoxides from sulfides using hydrochloric acid as a bifunctional reagent . This strategy accommodates a broad range of substrates and offers high diastereoselectivity and regioselectivity.
Several LSF modifications of amino acids and pharmaceutical derivatives further emphasized its utility. Mechanistic studies have demonstrated that the key to this selective chemical conversion lies in the dual oxidation process at the anode. The authors suggest that anodic oxidation of the sulfide generates a sulfur radical cation intermediate, which reacts with water at the anode to form a sulfoxide. Subsequent hydrogen atom abstraction by a chlorine radical leads to the formation of an intermediate carbon radical, whose resonant intermediate reacts with another chlorine radical to produce the desired α-chlorosulfoxide product (Scheme 14).
![[1860-5397-20-214-i14]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Electrochemical dual-oxidation enables access to α-chlorosulfoxides.
1.1.2 Unsaturated bond functionalization. Difunctionalizations of double and triple bonds are of high interest as they allow the introduction of two functional groups in a single step. An interesting electrochemical difunctionalization of styrene and cyclic olefin derivatives has been reported by the Hu group . They combined oxyformylation with brominations/chlorinations/trifluoromethylations using DMF and NaBr/NaCl/NaSO2CF3 as readily available reagents. The reported yields for this regio- and chemoselective transformation are high. For each reaction type, one LSF example was demonstrated using an estrone derivative. Mechanistically, this transformation can be understood as follows: first, a Br/Cl/CF3 radical is formed via anodic oxidation, which subsequently attacks the olefin. The newly formed benzyl radical is oxidized to a carbocation, which undergoes nucleophilic attack by DMF. Hydrolysis of the imine delivers the final product (Scheme 15).
![[1860-5397-20-214-i15]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Regio- and chemoselective formyloxylation–bromination/chlorination/trifluoromethylation of alkenes.
The synthesis of aziridines can be achieved via the formation of nitrenes in either a metal-catalyzed or metal-free fashion. In this context, Wickens and colleagues presented a remarkable dication pool strategy for accessing N-alkylaziridines via metastable dicationic intermediates derived from the interaction of non-activated alkenes with thianthrene . This procedure has the advantage of separating the oxidative activation of the alkenes from the aziridination step, allowing efficient access to a variety of aziridine building blocks containing sensitive functional groups. This was demonstrated by the LSF of primary natural and pharmaceutical amines carrying potential competing nucleophiles, such as tryptamine and primaquine (Scheme 16).
![[1860-5397-20-214-i16]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Aziridine formation by coupling amines and alkenes.
In the context of electrochemical difunctionalizations the Lei group published a transition-metal-free electrochemical difunctionalization method for the formation of C–S bonds . This method achieves the difunctionalization of a carbon atom by reacting isocyanide 2-isocyanoacetate with a thiophenol and an alcohol. The scope is very broad for thiophenols, alcohols, and isocyanides, and even alkyl thiols are compatible. In addition to aliphatic alcohols, benzyl alcohols are also suitable reagents. Numerous LSF examples and upscaling were demonstrated. The mechanism involves two anodic oxidations: first, the thiophenol is oxidized at the anode, forming a sulfur radical that attacks the isocyanide. The newly formed carbon radical is then oxidized to a carbocation, which is subsequently attacked by the alkoxide to furnish the final product (Scheme 17).
![[1860-5397-20-214-i17]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Formation of iminosulfide ethers via difunctionalization of an isocyanide.
The Lei group also demonstrated C–F-bond formations, particularly developing an electrochemical method for the cleavage of C–C bonds and the 1,3-difunctionalization of arylcyclopropanes . This electrochemical approach provides a convenient strategy for constructing 1,3-difluorinated molecules by employing Et3N·3HF as a nucleophilic fluorine source. Due to the mild reaction conditions, the LSF was demonstrated for complex natural precursors such as 5α-cholestan-3β-ol and androsterone scaffolds. During the reaction, the arylcyclopropane is oxidized at the anode to form a radical cation, causing the weakening of the Cα–Cβ bond. The radical cation then undergoes a three-electron SN2 reaction to generate a benzylic radical, which loses an electron at the anode to form a benzylic carbocation. Nucleophilic attack on the benzylic carbocation results in a 1,3-difunctionalized product (Scheme 18).
![[1860-5397-20-214-i18]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of 1,3-difunctionalized molecules via C–C-bond cleavage of arylcyclopropane.
The introduction of two heteroatoms was reported by Liu, Li, and Jin . They developed a method demonstrating excellent tolerance for a wide range of readily available alkenes and O,N-centered nucleophiles, showcasing 118 examples with good to high yields. The LSF of complex molecules, such as probenecid and estrone, highlighted the potential application of this method in the synthesis of selenium-containing drugs (Scheme 19).
![[1860-5397-20-214-i19]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Electrooxidative amino- and oxyselenation of alkenes. VBImBr = 1-butyl-3-vinylimidazolium bromide.
1.1.3 Annulation. Annulation refers to the formation of rings, a process that involves building a ring onto a preexisting system, whether cyclic or non-cyclic. The Lei group developed an electrooxidative annulation reaction that facilitates LSF . A regio- and stereoselective protocol was established for the [4 + 2] annulation of indole derivatives, allowing access to highly functionalized pyrimido[5,4-b]indoles due to its high functional group tolerance. Multiple examples were demonstrated with indole 1H-carboxamides linked to drug molecules or natural products at the R2 position. Additionally, an alkyl azide at the R2 position and an iodide at the R1 position were tolerated, enabling further functionalization. The proposed mechanism involves radical–radical cross-coupling. The indole 1H-carboxamide generates a nitrogen-centered radical during anodic oxidation in the presence of a base, while the 1,3-dimethylindole derivative forms an indole radical cation. The radical–radical cross-coupling between these two intermediates, followed by intramolecular cyclization and subsequent deprotonation results in the desired product (Scheme 20).
![[1860-5397-20-214-i20]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Electrooxidative dehydrogenative [4 + 2] annulation of indole derivatives.
Furthermore, Budny and coworkers demonstrated that (±)-triticonazole and related compounds could be cyclized and alkoxylated to the corresponding 1,2,4-triazolium tetrafluoroborates under electrochemical conditions . The reaction is conducted with a stoichiometric amount of HBF4, which converts the substrate to the corresponding cationic intermediate via a protonation, eliminating the need for an additional supporting electrolyte. The proposed mechanism involves the one-electron oxidation of triticonazole to form a radical cation, followed by cyclization to an intermediate. Subsequent anodic oxidation forms a doubly charged cation, which is then captured by methanol and deprotonated to yield the final product (pathway A). Additionally, due to the protonation of triticonazole, the participation of the protonated form in the overall reaction mechanism is also considered in pathway B (Scheme 21).
![[1860-5397-20-214-i21]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Electrochemical cyclization combined with alkoxylation of triticonazole.
Benzo[c][1,2]oxazines are useful scaffolds for the synthesis of natural products. In 2021, the Han group developed the electrochemical [4 + 2] annulation of hydroxamic acids 54 with alkenes for approaching benzo[c][1,2]oxazines . This method successfully achieved the LSF of several natural products such as lithocholic acid and estrone, affording the following benzo[c][1,2]oxazine derivatives in moderate to good yields (Scheme 22).
![[1860-5397-20-214-i22]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Electrochemically tuned oxidative [4 + 2] annulation of olefins with hydroxamic acids.
The cyclization of 2-ethynylanilines has been proven to be one of the most effective strategies for synthesizing indole derivatives. In this regard, Wang and coworkers developed the electrosynthesis of 3-iodoindoles from 2-ethynylanilines under mild and straightforward conditions . The functionalization of complex molecules, such as naproxen and cholesterol derivatives, demonstrated good functional group compatibility (Scheme 23a). In the same year, Wang and Huang group reported a similar approach using electrochemical methods to synthesize 3-selenylindoles via the cyclization of 2-ethynylanilines and diselenides (Scheme 23b) .
![[1860-5397-20-214-i23]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Electrosynthesis of indole derivatives via cyclization of 2-ethynylanilines.
1.2 Organo-mediators-enabled anodic oxidationThe Baran group has made significant contributions to the field of organic electrochemistry. One of their developments is the electrochemical allylic oxidation, a highly useful C–H functionalization method applicable to several natural products such as mono-, di-, tri-, and sesquiterpenes, along with some steroids . Crucial to their method was the use of a phthalimide-based mediator, adopted from earlier works (1968–1985). RVC electrodes with a highly developed surface area were employed for the reaction. The proposed mechanism is as follows: pyridine deprotonates tetrachloro-N-hydroxyphthalimide (R2N–OH), which is subsequently anodically oxidized. The resulting N-oxyl radical abstracts a hydrogen atom from the position adjacent to the olefin, forming an allylic radical. This allylic radical then reacts with cathodically generated tert-butyl peroxide to form an allylic peroxide, which ultimately transforms into an enone upon elimination of t-BuOH (Scheme 24).
![[1860-5397-20-214-i24]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Allylic C–H oxidation of mono-, di-, and sesquiterpenes.
One year later, they developed an electrochemical transformation closely related to their electrochemical allylic oxidation, i.e. the oxidation of unactivated C(sp3)–H bonds (Scheme 25a) . Besides, the same group published a comprehensive analysis on N-ammonium ylide mediators, which were found to be superior to quinuclidine scaffolds for a chemoselective C(sp3)–H oxidation (Scheme 25b) .
![[1860-5397-20-214-i25]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Oxidation of unactivated C–H bonds.
The electrochemical C(sp3)–H fluorination of unactivated C–H bonds is another important transformation via anodic oxidation realized by the Baran group . The choice of Selectfluor, which plays multiple roles, was crucial. In addition to functioning as a fluorine source, Selectfluor also acts as a mediator similar to quinuclidine and serves as an electrolyte. The method was demonstrated to be scalable for natural products such as sclareolide and protected ʟ-valine. The proposed mechanism involves a radical chain process. Initiation occurs by nitrate-mediated or direct electrochemical anodic oxidation, followed by fluorination with Selectfluor. After fluorination, the Selectfluor reagent abstracts a hydrogen atom from the substrate, which can then undergo further fluorination. The nitrate additive proved helpful as an initiator but is not necessary for certain substrates like sclareolide and protected ʟ-valine (Scheme 26).
![[1860-5397-20-214-i26]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Fluorination of C(sp3)–H bonds. rAP = rapid alternating polarity.
An electrochemical C(sp3)–H cyanation for LSF was reported by the Stahl group . This protocol relies on the sterically encumbered ABNO (9-azabicyclononane N-oxyl) as a mediator, with TMSCN serving as the cyanide source. The reaction operates at low potentials, resulting in high functional group tolerance, even accommodating secondary alcohols. Additionally, pyrrolidine, anazepane, and morpholine scaffolds successfully underwent the reaction. Another notable feature of this method is its high diastereoselectivity. All products were ultimately obtained as p-toluenesulfonic acid salts (Scheme 27).
![[1860-5397-20-214-i27]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: C(sp3)–H α-cyanation of secondary piperidines.
In 2021, Zhang et al. developed an electrochemical method for the hydrolysis of hydrosilanes to silanols using N-hydroxyphthalimide (NHPI) as the hydrogen-atom-transfer (HAT) mediator . To demonstrate the potential of their approach, they showcased the LSF of natural products such as (−)-borneol and (+)-fenchol, as well as pharmaceutical drugs including ibuprofen, febuxostat, and gemfibrozil, achieving moderate to good yields. The proposed mechanism involves the oxidation and deprotonation of NHPI at the cathode to form phthalimide-N-oxyl (PINO) radicals. These PINO radicals act as HAT reagents, abstracting a hydrogen atom from the Si–H bond of the hydrosilane to generate a silyl radical. This silyl radical is then oxidized anodically to produce a silyl cation. The silyl cation subsequently abstracts a proton from water (H2O), forming the desired silanol product (Scheme 28).
![[1860-5397-20-214-i28]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Selective electrochemical hydrolysis of hydrosilanes to silanols.
While many methods combining metal catalysis and electrochemistry have been developed, the combination of electrochemistry with organocatalysis is generally less explored. In this context, Wang et al. combined organocatalysis and electrochemistry for the benzyl amination via C–H/N–H dehydrogenative cross-coupling of alkyl arenes with azoles . According to the authors, the reaction proceeds via hydrogen-atom transfer (HAT) at the benzylic position, mediated by DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone). The proposed mechanism includes two possible pathways: In path A, the benzylic position undergoes HAT to form a benzyl radical, which is then oxidized by the DDQH• radical to generate a carbocation and DDQH−. In path B, the reaction involves direct hydride transfer to DDQ, forming DDQH− and a carbocation. In both pathways, the amine nucleophile captures the carbocation, resulting in the final amination product after losing a proton. Subsequently, DDQH− is protonated to produce DDQH2. The anodic oxidation of DDQH2 regenerates DDQ, which re-enters the catalytic cycle (Scheme 29).
![[1860-5397-20-214-i29]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-214-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Organocatalytic electrochemical amination of benzylic C–H bonds.
Furthermore, Qiu and coworkers disclosed a metal-free electrochemical dihydroxylation of unactivated alkenes using water as the hydroxy source under air conditions .
This mild method proceeds with a broad range of unactivated
Comments (0)