
Remember me




Predispositions to germline mutations have long been recognized as significant contributors to the development of various solid tumors [1]. Leukemia susceptibility is primarily associated with clinical phenotypes, such as dyskeratosis congenita (DC), down syndrome, and fanconi anemia (FA). However, in the last 10 years, population and family studies have identified several germline genetic mutations that increase the risk of leukemia in carriers [1]. Myeloid neoplasms associated with germline predisposition, including acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), occur more frequently in individuals with genetic conditions associated with an increased risk of myeloid malignancies [2]. These diseases are now grouped using a conventional approach that couples the myeloid disease phenotype with the predisposing germline genotype; for example, AML with germline pathogenic variants in DDX41. Genetic counseling and the evaluation of family history are integral components of the diagnostic evaluation of index patients [3].
The recent inclusion of the ‘Myeloid Neoplasms with Germline Predisposition’ category in the revised fifth edition of the World Health Organization (WHO) Classification of myeloid and histiocytic/dendritic neoplasms emphasizes the growing recognition and importance of germline evaluation in patients with myeloid neoplasms [4]. Based on the high frequency of deleterious germline variants identified in patients aged < 40 years with AML and MDS, all such patients should undergo germline genetic testing at disease onset, regardless of age, family history or syndromic features [5,6,7,8,9].
Identification of hematologic neoplasms with germline mutations is critical for accurate diagnosis, patient management, screening of related donors for stem cell transplantation, selection of therapeutic strategies, and genetic counseling of affected family members. A high index of suspicion for germline mutations is important, particularly for younger patients diagnosed with hematologic neoplasms and those slated for transplantation using related donors. The unwitting use of related transplant donors harboring the same germline mutation as the patient has resulted in donor-derived AML or MDS leading to poor outcomes, underscoring the need for increased awareness and recognition of myeloid neoplasms with germline predispositions [10].
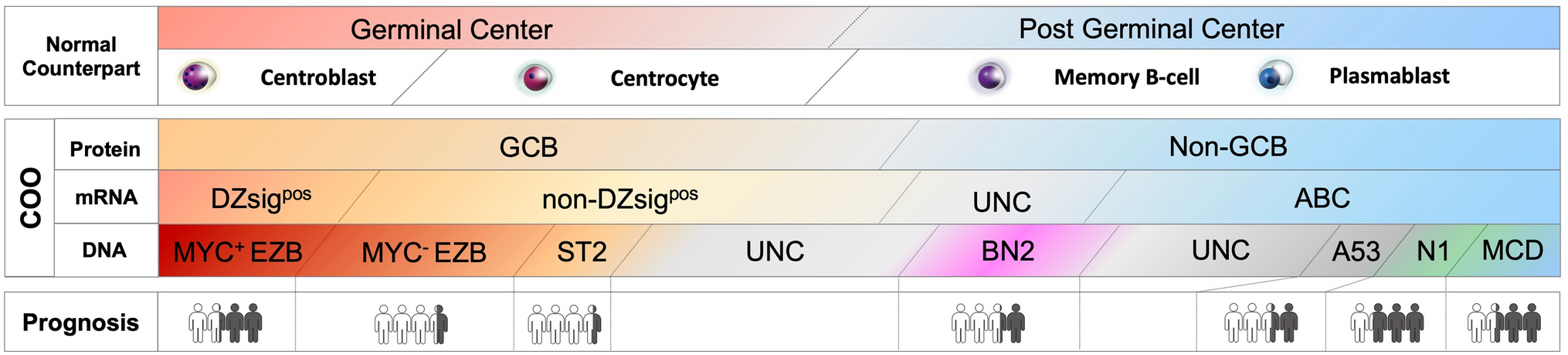
Subtypes of myeloid neoplasms with a germline predispositionThe clinical manifestations of myeloid neoplasms are grouped into three subtypes in the fifth Edition of the WHO classification (Fig. 1, Table 1), to which most germline predisposition conditions can be assigned. These subtypes are classified based on: (1) systemic organ abnormalities and (2) thrombocytopenia. For instance, cases with no systemic organ abnormalities or thrombocytopenia are categorized as “myeloid neoplasms with germline predisposition without a pre-existing platelet disorder or organ dysfunction.” In contrast, cases with thrombocytopenia are classified as “myeloid neoplasms with germline predisposition and a pre-existing platelet disorder,” while those with systemic organ dysfunction fall under the category “myeloid neoplasms with germline predisposition and a potential organ dysfunction.” In addition, the International Consensus Classification (ICC) has introduced a new classification based on the 2016 WHO classification, and incorporated updated clinical factors and genetic abnormalities [10]. The ICC has identified hematopoietic neoplasms with a germline predisposition, which includes not only myeloid malignancies, but also lymphoid malignancies.
Fig. 1
Subtypes of myeloid neoplasms with germline predisposition. Myeloid neoplasms with germline predispositions are classified according to (1) systemic organ dysfunctions and (2) thrombocytopenia. These three subtypes are characterized by different genes
Table 1 Common germline predispositions in acute myeloid leukemiaMyeloid neoplasms with germline predisposition without a pre-existing platelet disorder or organ dysfunctionThis category includes genes such as DDX41, which predisposes patients to both myeloid and lymphoid neoplasms, and CEBPA, which predisposes patients to AML. Moreover, TP53 has been included in this category, recognizing the importance of Li-Fraumeni syndrome and its predisposition to myeloid and lymphoid malignancies in both treatment-naïve and therapy-related settings [11, 12].
Myeloid neoplasms with germline DDX41 mutationsThe gene DDX41 is located at the distal end of the long arm of chromosome 5 (5q35.3). It encodes a DEAD-box RNA helicase that is involved in RNA splicing, immune responses, and ribosomal biogenesis. DDX41 germline mutations are the most common genetic predispositions for myelodysplastic syndrome and AML [13, 14].
Makishima et al. examined DDX41-associated susceptibility to myeloid neoplasms which occurred at a frequency of 3.8% (346/9082) [15]. This phenotype is related to heterozygous germline mutations in the DDX41 and does not manifest clinical phenotype until disease progression. The acquisition of a somatic mutation in the second DDX41 allele typically leads to MDS or AML, commonly occurring in the sixth decade of life, similar to the age of onset of typical sporadic diseases. Mutant DDX41 carriers have a lifelong risk of developing myeloid malignancy of approximately 50%, with a higher frequency of disease progression in men than in women. Patients with MDS with DDX41 mutations have a high risk of rapidly progressing to AML, particularly those with truncating variants [16].
Interestingly, patients with germline DDX41 mutations develop AML or MDS in adulthood, in contrast with most other constitutional disorders that typically appear in childhood. In fact, the first described family with DDX41 mutations consisted of a father in his 70 s and a brother and sister in their 40 s [17]. Furthermore, there is an estimated male to female predominance of 3:1 among carriers of myeloid tumors. The reported germline alterations are frameshift mutations, with the D140fs mutation being the most common [16]. Known germline alleles are associated with different ethnicities, including preliminary data suggesting A500fs mutation in Asian populations. Somatic missense mutations in DDX41 occur frequently in the second allele of patients with germline DDX41 loss-of-function mutations, with DDX41 R525H being the most prevalent [4].
Two distinct DDX41 somatic mutations in both alleles predicts leukemic transformation much better than the revised and molecular International Prognostic Scoring Systems (IPSS-R and IPSS-M) [18, 19]. Considering that virtually all truncating variants are germline, the identification of a truncating DDX41 allele via genomic profiling for the detection of somatic mutations should prompt germline genetic testing. Finally, patients with deleterious germline DDX41 variants frequently develop severe acute graft-versus-host disease following allogeneic stem cell transplantation unless they receive post-transplant cyclophosphamide [15]. This observation suggests the presence of a pro-inflammatory milieu that stimulates donor-derived T cells [16].
Myeloid neoplasms with germline CEBPA mutationsPatients with familial AML with mutated CEBPA typically have no other hematopoietic or systemic manifestations before AML development. AML occurs in nearly all patients between the ages of 2 and 59 years who carry these mutations. Leukemia usually exhibits a normal karyotype, frequently with Auer rods, and demonstrates an aberrant expression of CD7 [20, 21].
AMLs with biallelic mutations in CEBPA (which encodes CCAAT/enhancer binding protein-α) are recognized as a unique subtype, demonstrating a more favorable outcome. Approximately 10% of these cases also harbor a germline CEBPA mutation, which leads to the development of AML with near-complete penetrance, typically in the third decade of life, and can occur without preceding MDS or the cytopenic phase [22, 23]. These germline mutations are commonly frameshift or nonsense mutations located at the N-terminus. AML progression is frequently associated with acquired mutations in the wild-type CEBPA allele [23, 24]. Somatic mutations in CEBPA are enriched in the basic leucine zipper domain (b-ZIP), which is a strong indicator of a higher chance of complete remission, improved overall survival, and a lower risk of relapse [25]. Notably, when these patients experience disease recurrence after chemotherapy, they develop new clones with a different spectrum of acquired mutations, including new somatic CEBPA mutations, demonstrating that these secondary leukemias are not true relapses [23]. Discordant somatic CEBPA mutations at the time of AML diagnosis and relapse have led to the identification of a distinct model of AML recurrence. Four out of six familial patients with AML with germline CEBPA mutations acquired somatic CEBPA mutations in different locations within the b-ZIP region upon relapse, compared to those present at diagnosis. One patient acquired a somatic N-terminal mutation, while another reverted to the wild-type sequence at relapse [23]. Patients with AML and germline CEBPA mutations have favorable outcomes, yet they are prone to secondary leukemias that remain responsive to chemotherapy, unlike true relapsed disease [4].
Myeloid neoplasms with germline TP53 mutationsTP53 is one of the most frequently mutated genes in cancer, particularly in adult-onset cancers. Germline mutations are the defining features of Li–Fraumeni syndrome (LFS) and predispose individuals to a diverse range of tumors, particularly sarcomas, central nervous system tumors, adrenocortical carcinoma, and breast cancer, as well as myeloid and lymphoid leukemia [4, 11].
LFS confers a lifetime risk of cancer, including leukemia, in approximately 70% of men and 100% of women. Leukemia is a core cancer associated with LFS, accounting for 3–6% of all LFS tumors [26]. Virtually all hematological malignancies are represented in the International Agency for Research on Cancer TP53 database, including therapy related MDS and AML [27]. Whether patients with LFS have lower remission rates with therapy remains to be determined; however, somatic TP53 mutations are associated with poor outcomes in both childhood and adult leukemia [28,29,30]. Diagnosing LFS or other cancer predisposition syndromes is essential, not only as it relates to genetic counseling and donor selection, but also in considering tumor surveillance studies after the successful treatment of a primary tumor [1].
Myeloid neoplasms with germline predisposition and a pre-existing platelet disorderPlatelet disorders are a group of blood disorders with variable severity and clinical impacts. Pathogenic germline variants, especially RUNX1, ETV6, and ANKRD26, are also associated with the risk of developing hematological malignancies. Although they may initially present with mild to moderate thrombocytopenia, each of these three disorders has a distinct penetrance of hematological malignancy and a different range of somatic alterations associated with malignancy development [14].
Myeloid neoplasms with germline RUNX1 mutationsFamilial platelet disorder with a propensity for myeloid malignancies (Online Mendelian Inheritance in Man [OMIM] 601,399), caused by pathogenic variants of RUNX1, is associated with variable degrees of thrombocytopenia, a functional platelet defect, and an increased risk for hematologic malignancies.
Germline mutations in RUNX1, first described in 1999 by Song et al. [31], result in the loss of RUNX1 protein function through point mutations that produce proteins acting as dominant negatives as well as nonsense or frameshift mutations and larger deletions [32]. The overall lifetime risk of progressing to myeloid disease is estimated at 44%, and it typically manifests during adulthood. This risk is particularly higher in patients with missense mutations, potentially due to the greater ability of the resulting proteins to inhibit transactivation of wild-type RUNX1 through dominant negative activity compared with loss-of-function alleles [32, 33]. However, eventual progression to a frank neoplasm is associated with the acquisition of somatic mutations in the remaining wild-type RUNX1 allele, as well as GATA2 mutations [34], and, less commonly, other genes recurrently mutated in AML and MDS, such as FLT3 (which encodes a receptor tyrosine kinase), DNMT3A (which encodes a DNA methyltransferase), KRAS, and splicing factors. Notably, patients with familial platelet disorders and a predisposition to myeloid malignancy also exhibit a higher incidence of clonal hematopoiesis at a younger age [35].
Myeloid neoplasms with germline ETV6 mutationsThrombocytopenia 5 (THC5; OMIM 616216) is an autosomal dominant syndrome caused by the inheritance of monoallelic mutations in the ETV6 gene, which typically presents with moderate thrombocytopenia with or without a mild bleeding propensity, hypo-lobulated megakaryocytes, and is associated with hematologic and other malignancies [36].
ETV6 is a member of the Erythroblast Transformation Specific transcription factor family, which is required for bone marrow (BM) hematopoiesis and megakaryopoiesis, as demonstrated in mouse KO models [37, 38]. ETV6 predominantly functions in conjunction with SIN3A, N-COR, and HDAC3 as a transcriptional repressor [39]. For ETV6, most of the pathogenic variants reside within its ETS domain, which is required for DNA binding, leading to a dominant-negative effect via the loss of transcriptional repression [40, 41]. Most variants are missense, although nonsense, frameshift, deletion, and structural variants leading to loss-of-function have also been observed [1, 4]. Etv6 megakaryocyte-erythroid progenitor conditional KO mice show thrombocytopenia due to an increase in megakaryocytic colony-forming cells, suggesting a deterioration in megakaryocyte maturation with the loss of Etv6 [42]. Interestingly, ETV6 interacts with the FLI1 oncoprotein to decrease its transcriptional activity. FLI1 is a platelet and megakaryocyte ETS domain transcription factor that has been implicated in hereditary thrombocytopenia [36].
ETV6 most frequently results in acute lymphoblastic leukemia (ALL), commonly B-ALL, with occasional occurrences of biphenotypic leukemia, chronic myelomonocytic leukemia, myeloma, and MDS/AML [43]. In childhood B-ALL cohorts, pathogenic germline ETV6 variants were identified in 0.8% of cases, typically in older children with hyperdiploid karyotypes [44]. Penetrance is incomplete, with only ~ 30% of carriers developing HM; however, the penetrance of thrombocytopenia is > 90% [36].
Myeloid neoplasms with germline ANKRD26 mutationsThrombocytopenia 2 (THC2; OMIM 188000) is an autosomal dominant disorder caused by ANKRD26 mutations that presents with moderate thrombocytopenia (with or without a mild bleeding propensity), dysmegakaryopoiesis, and the development of hematologic malignancies in adulthood [45].
In ANKRD26-related thrombocytopenia (ANKRD26-RT), the bleeding risk is fairly low, and thrombocytopenia tends to be moderate with normal platelet size and no consistent defect in in vitro aggregation studies. Careful examination of cases and family histories indicates that patients with ANKRD26-RT are at increased risk of MDS and leukemia [46, 47]. However, the risks of lymphoid malignancy and nonhematologic cancers do not appear to be increased [1].
There is an estimated 8% lifetime risk of progression to MDS, AML, or chronic myeloid leukemia [45,46,47,48]. In addition, some patients develop erythrocytosis and leukocytosis [47, 49]. In a recent study on a pediatric cohort with germ line mutations in RUNX1, ETV6, or ANKRD26, mild-to-moderate thrombocytopenia and mild bleeding phenotypes were identified. Hematologic malignancies also coexisted with RUNX1 (10/14) and ETV6 (1/2) variants but not with ANKRD26 (0/8 patients), consistent with the reduced penetrance of ANKRD26 for hematologic malignancies [36, 50].
Myeloid neoplasms with germline predisposition and potential organ dysfunctionSimilar to the 2016 WHO classification, this category contains germline mutations in GATA2, germline mutations associated with representative BM failure disorders, germline mutations affecting RAS pathway genes (NF1, PTPN11, and CBL) associated with neurofibromatosis, Noonan-like syndromes predisposing to JMML, and Down syndrome, which predisposes to both myeloid and lymphoid neoplasia [10]. Additions to this group include SAMD9 and SAMD9L, with both predisposing individuals to acquired monosomy 7/del(7q) and MDS, respectively [6, 51,52,53].
Myeloid neoplasms with germline GATA2 mutationsGATA2 mutations have a heterogeneous presentation and may be associated with MDS, AML, aplastic anemia, congenital neutropenia, or other hematological abnormalities [54, 55]. It is one of the most commonly inherited AML/MDS disorders, particularly in children and young adults with MDS [56, 57].
In addition to MDS and/or AML, patients may also have immunodeficiency, sensorineural hearing loss, lymphedema, and dermatological or pulmonary manifestations [55]. Immunodeficiency associated with infectious pathogens, such as human papillomavirus and nontuberculous mycobacteria, has resulted in recommendations for early human papillomavirus vaccination and antibiotic prophylaxis. Germ line mutations in GATA2 are also found in approximately 7% of all children with MDS and in approximately two-thirds of adolescents with monosomy 7 MDS, making it the most common germline defect predisposing pediatric MDS [54]. Surveying GATA2 in children with MDS is crucial, even in the absence of a family history, as it can inform supportive care strategies and/or accelerate planning for hematopoietic stem cell transplantation [1]. Inherited GATA2 deficiency is associated with severe deficiency of B lymphocytes, monocytes, NK cells, and dendritic cells, leading to cytopenia of these lineages and subsequent infections. The inheritance pattern of GATA2 deficiency is autosomal dominant [58]. The acquisition of cytogenetic abnormalities (monosomy 7 or trisomy 8) or somatic gene mutations (primarily in STAG2 or ASXL1) may cause progression to myeloid neoplasms, more commonly MDS and less frequently AML, or chronic myelomonocytic leukemia. Allogeneic transplantation in GATA2 deficiency reverses the hematological disease phenotype [59]. However, owing to the highly variable clinical course of GATA2 deficiency, preemptive transplantation is not recommended. Rather, proper surveillance and expectant management [16].
Myeloid neoplasms with germline SAMD9/SAMD9L mutations (MIRAGE syndrome)Inherited mutations in SAMD9 or SAMD9L are common causes of MDS in children, especially pediatric bone marrow failure/MDS with monosomy 7. These patients develop bone marrow failure, MDS alone, and/or a wide variety of somatic abnormalities. Homozygous deletions of SAMD9 result in normophosphatemic familial tumor calcinosis (OMIM 610455), characterized by the deposition of calcified tumors, whereas heterozygous mutations (OMIM 617053) cause MIRAGE syndrome, characterized by myelodysplasia with monosomy 7, infection, growth restriction, adrenal hypoplasia, genital phenotypes, and enteropathy [60]. Heterozygous mutations in SAMD9L (OMIM 159550 and 611170) cause ataxia, pancytopenia, and monosomy 7 MDS [60].
Germline mutations in SAMD9 or SAMD9L, two interferon-inducible genes located on chromosome 7, were initially found to cause distinct multisystem syndromes characterized by neurological and/or endocrine abnormalities, as well as MDS with monosomy 7 [51, 60,61,62,63]. Recent studies have shown that germline mutations in these genes can also be found in isolated and familial pediatric MDS and AML [
Comments (0)