
Remember me
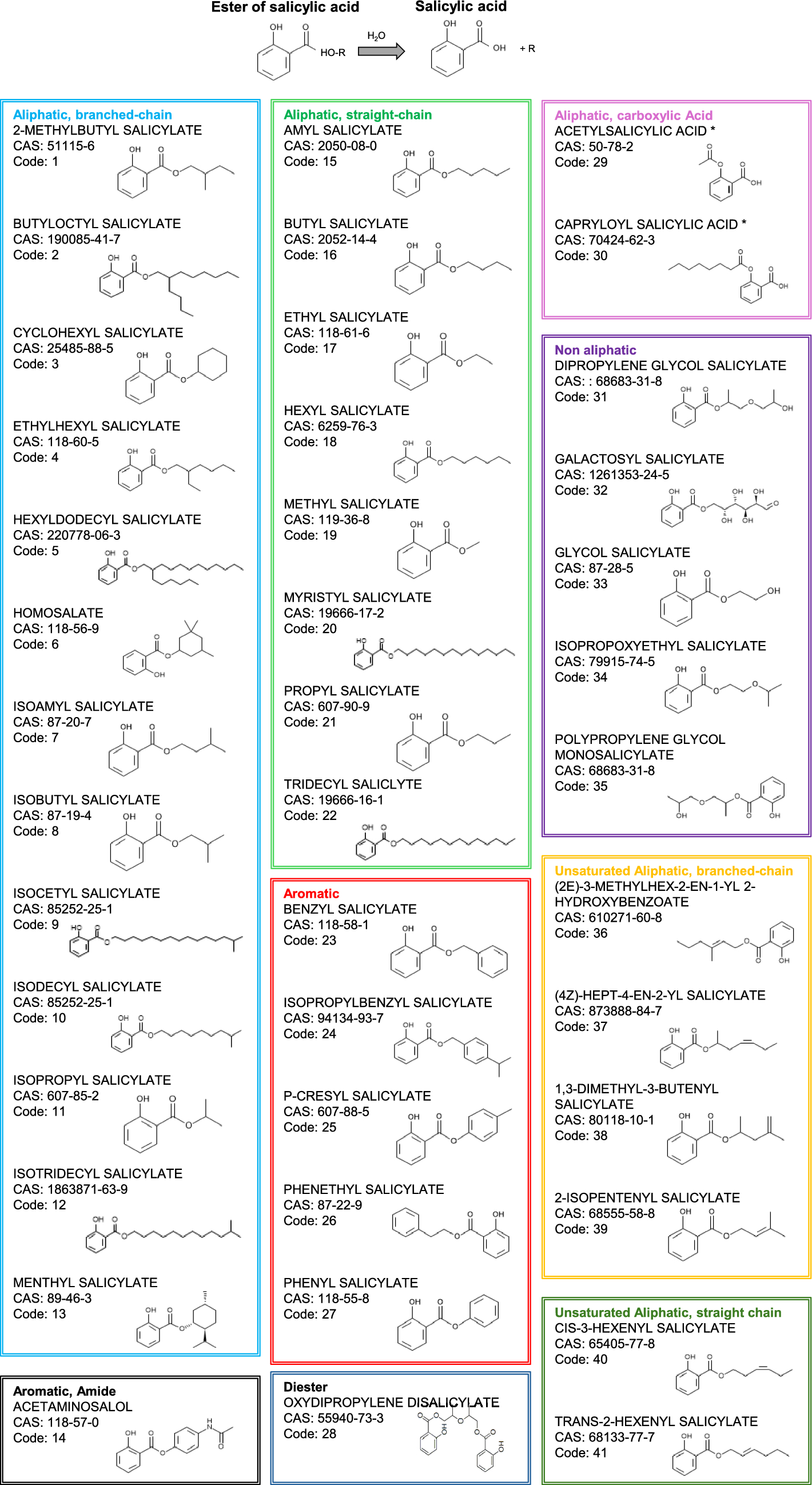
To detect gene expression changes upon okadaic acid (OA) treatment for 24 h in HepaRG cells, we performed RNA sequencing and differential gene expression (DGE) analysis. As described in the Materials and methods section, raw counts were filtered resulting in 22,122 genes expressed above the threshold. After normalization and transformation of processed counts data, Principal Component Analysis (PCA) was used to visualize and reduce the complexity of the dataset. Figure 1A shows the PCA scores plot for all twelve samples analyzed by RNAseq. PC1 and PC2 representing most of the variance clearly separate the samples treated with an increasing concentration of OA from the negative control. This is also reflected by the number of differentially expressed genes (DEGs) that peaks at 4200 down- and 3586 up-regulated upon treatment with 100 nM OA (Fig. 1C). We found that 184 and 513 genes were significantly down- and up-regulated in common between all concentrations of OA treatment, respectively (Supplemental Fig. 1). Overall, there were 8181 genes that were significantly changed in their expression in at least one condition which reflects 36.9% of the studied genes (Supplemental Table S5).
Fig. 1
Global omics data results for transcriptomics and proteomics. Scores plots of principal component analysis (PCA) for transcriptomics (A) and shotgun proteomics (B). Colors indicate the different treatments: negative control = 0 nM OA (gray), 11 nM OA (light green), 33 nM OA (green), 100 nM OA (dark green) for transcriptomics; incubation time of 0.5 h (light blue), 4 h (blue), 24 h (dark blue) and negative control (circle), 33 nM OA (square), 100 nM OA (diamonds) for proteomics. Bar plots for number of significantly changed genes (C) and proteins (D). Genes with padj < 0.1 and |log2FC|> 1 were considered as significant and proteins with padj < 0.05 and |log2FC|> 0.5. Venn diagrams comparing significantly regulated genes/proteins for treatment with 33 nM (E) and 100 nM OA (F)
In our study, GO term enrichment analysis was performed to identify significant biological processes affected by different concentrations of OA. In summary of the dot-plot in Fig. 2, the analysis highlighted that genes up-regulated at 33 nM were predominantly associated with cell cycle regulation and DNA replication. At higher concentrations (100 nM) also GO terms related to reactive oxygen species (ROS) and apoptosis signaling pathways were associated (Fig. 2). Down-regulated genes were consistently enriched for fatty acid and xenobiotic metabolism for both OA concentrations. The underlying CAR signaling pathway is depicted for OA 33 nM treatment based on top “canonical pathways” results from Ingenuity Pathway Analysis (IPA) in Fig. 3 with key regulators, such as RXRA, NCOA, and CAR (NR1I3) itself showing decreased gene expression. CAR-regulated targets, including CYPs, UGTs, and transporters (e.g., SLCO1B1, ABCC3), are also down-regulated leading to an inactivation of the downstream xenobiotic metabolism.
Fig. 2
Dot-plot of enriched GO terms for transcriptomics (RNAseq) and Shotgun proteomics. Only results for OA treatment (33, 100 nM) after 24 h are depicted and split by up-/down-regulated genes/proteins. The color indicates the significance measured by the adjusted BH p value and the dot size indicates the gene ratio per GO term
Fig. 3
Overview of xenobiotic metabolism regulated by the CAR signaling pathway from Ingenuity Pathway Analysis (IPA). Transcriptome data (log2FC values) for treatment with 33 nM OA (24 h) were mapped while results for 100 nM OA (24 h) showed a similar picture (not shown). CAR is labeled by its synonym NR1I3. Genes detected in our dataset are shown in green (down-regulated) or red (up-regulated). Genes shown in orange or blue represent a predicted up- (orange) or down-regulation (blue)
Shotgun proteomicsIn addition to RNA sequencing, we also carried out Shotgun Proteomics analysis to identify proteins that were affected by OA (33 and 100 nM) in their expression. As the transcriptomic analysis focused exclusively on the changes in signaling pathways after 24 h, we wanted to include the relevant protein changes at earlier time points (0.5 and 4 h) as well. After processing and filtering the dataset as described in the Materials and methods section, 3028 unique proteins were detected and quantified. The PCA scores plot for all 27 samples analyzed by Shotgun Proteomics, depicted in Fig. 1B, clusters the samples along PC1 and PC2 based on factors of concentration and time. The samples of each time point are separated by the increasing OA concentration, as the samples of 100 nM are more distant from the negative control than those of 33 nM. The treatment effects on the proteome for 0.5 and 4 h seem to be more similar to each other as the samples are clustering together, while those for 24 h treatment were separated.
Subsequently, each OA treatment was compared to the negative control of the respective time point using Student’s t test, and significant protein changes were considered for adjusted p values < 0.05 and |log2FC|> 0.5. In general, the percentage of differentially expressed proteins (DEPs) was 31.3% (947 proteins changed in at least one condition), which is quite similar to the mRNA level even if the absolute numbers are lower.
In agreement with the PCA scores plot, the concentration-dependent effect on the number of DEPs is also indicated in Fig. 1D as already observed for DEGs (Fig. 1C). When comparing the DEGs identified by RNAseq with deregulated proteins, we found an overlap of 71 and 173 molecules for treatment with 33 and 100 nM OA, respectively (Fig. 1E–F). For instance, SERPINB2, ITGA2, and SLC1A5 were commonly up-regulated, while CYP2E1, FABP1, and ADH1B were down-regulated at OA both levels. Interestingly, the extent of up- or down-regulation seemed to be higher at the transcriptomic than at the proteomic level which is apparent from the heatmaps in Supplemental Fig. 2. Indeed, correlation analysis of log2FC values (100 nM OA) of both omics levels results in a correlation coefficient of 0.79 and a slope of 2.38 for linear model of transcriptomics in dependence of proteomics. Also, for 33 nM OA, a factor > 2 was derived for this significant relationship, as summarized in Supplemental Fig. 3. Besides this set of commonly regulated molecules, each level showed specifically deregulated genes or proteins that were not identified by the other method.
Next, we analyzed the overlaps between sets of DEPs and visualized them in Venn diagrams. For both OA concentrations, they indicate rather distinct proteomic changes at the early stage (0.5 and 4 h) and late stage (24 h) as there are many specific proteins at each condition and few common DEPs (Supplemental Fig. 4).
Fig. 4
Boxplot of log2FC across eight clusters of DEPs measured by shotgun proteomics over time and per OA concentration (33 nM in green, 100 nM in dark green) according to kmeans clustering
To get a better understanding of the biological pathways affected by OA treatment over time, we performed kmeans clustering analysis to group all 947 DEPs into eight clusters. The boxplots in Fig. 4 show the different concentration- and time-dependent proteomic changes and the enriched GO terms associated with each cluster are summarized in Supplemental Table 2. Cluster 2 contains 100 proteins, such as CYP2E1, ABCC3, and FABP1, that are mainly down-regulated after 24 h and enriched for GO terms related to xenobiotic and lipid metabolism (Supplemental Fig. 5A). However, cluster 4 reflects an early proteomic response including 155 down-regulated proteins that are associated with ribosome biogenesis and translation, e.g., RRP8, RPL28, and EIF5B. Multiple clusters indicate different patterns of up-regulated proteins, as clusters 1 and 7 are reaching the maximum expression after 24 h, while clusters 3, 5, and 6 are showing early proteomic changes upon OA treatment. Clusters 3, 5, and 6 are mainly enriched for biological processes like RNA splicing and fatty acid metabolism; on the other hand, proteins in cluster 1 and 7 are associated with apoptosis and mRNA stabilization, respectively. Moreover, proteins involved in actin filament organization appear in cluster 6 (early up-regulation, e.g., CTNNA2, PAK1, and SPTBN4) and 7 (late up-regulation, e.g., MARCKS, STMN1, and ZYX), as summarized in Supplemental Fig. 5B.
Fig. 5
Distribution of log2FC ratios for OA treatment for 0.5, 4 and 24 h in the phosphoproteome dataset. The left side shows the density plot for 33 nM OA (OA33) and the right side for 100 nM OA (OA100)
PhosphoproteomicsAs a potent phosphatase inhibitor of mainly PP1 and PP2A, effects of OA on the phosphorylation status of the cells were expected. To investigate the signaling events associated with OA treatment and subsequent PP1 and PP2A inhibition, we performed LC–MS-based profiling of enriched phosphopeptides. In total, we detected 288 unique phosphopeptides that were used as input for a PCA plot that shows a clear separation of samples according to concentration and duration of the OA treatment (Supplemental Fig. 6A). To distinguish changed phosphoprotein abundances from altered phosphorylation status, we performed a pairwise normalization of enriched phosphopeptides relative to their counterparts in the non-enriched shotgun proteomics dataset. From the total number of 288 phosphopeptides, 72 could be matched to peptides from the shotgun data which resulted in log2FC ratios.
Fig. 6
Boxplot of log2FC ratio across five clusters of phosphopeptides over time and per OA concentration (33 nM in green, 100 nM in dark green) according to kmeans clustering
Strikingly, OA treatment results in a global increase in phosphorylation status as indicated by the shift of log2FC ratios to the positive values in Fig. 5. This effect is even more pronounced for treatment with 100 nM OA compared to 30 nM with median values of 0.631 and 0.490, respectively (see Supplemental Table 3 for details). This observation is also reflected by a higher number of phosphopeptides with increased log2FC ratios than decreased values (Supplemental Fig. 7A) peaking after 4 h of OA treatment.
Fig. 7
Phosphoproteomic changes connected to actin cytoskeleton. Heatmap of selected phosphopeptides related to actin filament organization (A). Relevant changes with |log2FC ratio|> 1 are indicated by “ + ” sign. Prior knowledge-based signaling network based on protein–protein interactions (PPIs) and kinase information from OmniPath for selected proteins (B). These proteins of interest (POI) were selected based on GO term annotation related to actin filament binding and organization. Additionally, phosphatases PP1 and PP2A (“primary targets”) as well as intermediate kinases are included in the network, and red edges indicate (de)phosphorylations in contrast to PPIs
To get a better understanding of the patterns in the phosphoproteome data, we analyzed the 72 matched phosphopeptides by kmeans clustering (Fig. 6). The five resulting groups reflect similarly reacting phosphopeptides upon OA treatment, such as cluster 1 containing 24 molecules (e.g., ADD3, PLEC, and SPTBN1) that accumulated phosphorylations after 0.5 h and 4 h, but this effect reversed after 24 h. Cluster 2 is also showing an increased phosphorylation status but not immediately, rather after 4 or 24 h with proteins like MTDH, NDRG2, and EIF4B. The biggest increase was observed for FKBP5 and USP5 in cluster 4. Across the entire dataset, there is a striking overrepresentation of proteins associated to actin filament organization, microtubule, and cadherin binding, highlighting a broad impact of OA treatment beyond the specific clusters identified.
Based on GO term annotation related to actin cytoskeleton, a subset of genes was selected for a more detailed inspection. Especially, after 4 h and 24 h, OA treatments lead to a pronounced increase of phosphorylation for PALLD, PLEC, AFDN, MARCKS, AHNAK, and SEPTIN9 (Fig. 7A). To get a better understanding of the signaling network around the proteins of interest existing knowledge for protein–protein interactions (PPIs) and kinase–substrate relationships from OmniPath utilized. The derived network in Fig. 7B shows the interactions between the primary OA targets PP1 and PP2A, selected proteins of interest (POI) and kinases as intermediate regulators. Among the most connected kinases in the network, we identified MAPK1 (ERK2), MAPK3 (ERK1), AKT1, CDK1, CDK2, GSK3B, TP53, PRKCA, and PRKCB as potential key regulators for POI that are associated with downstream effects in the actin cytoskeleton organization. Then, we explored phosphopeptides related to microtubule and cadherin binding that partially overlapped with POI for actin binding. Additionally, we recognized NDRG1 and DYNC1I2 as phosphorylated POI for microtubule organization and CTNND1 and PPL for cadherin binding/focal adhesion (Supplemental Fig. 8).
Fig. 8
Overview of DigiWest results. Boxplot of ratio across six clusters of 136 analytes measured by DigiWest over time and per OA concentration (A). Venn diagram comparing the unique proteins detected by Shotgun Proteomics, Phosphoproteomics, and DigiWest (B)
DigiWest (targeted proteins)In a targeted approach, we used the targeted DigiWest method to complement the previous untargeted (phospho)-proteomic methods, covering 132 analytes that correspond to 97 unique proteins (some with different modifications). Fig. 8B illustrates that the majority of those proteins (63%) could exclusively be detected by DigiWest. Similar to the phosphoproteome data, samples cluster according to OA concentration and treatment duration in the PCA scores plot (Supplemental Fig. 6B). This is also exemplified by the rising number of down- or up-regulated analytes (|log2FC|> 1) with increasing OA concentration and over time (Supplemental Fig. 7B). However, we observed an early regulation at the proteome level already 0.5 h after OA treatment, which is in agreement with the Shotgun Proteomics data (Fig. 1D).
Kmeans clustering was applied to group similarly behaving analytes into groups as summarized by boxplots in Fig. 8A. Cluster 2 and 5 show strong accumulation upon OA treatment of 12 and 24 h, especially for 100 nM. Among them are the phosphorylated proteins RPS6, JUN and RB (cluster 2) as well as MAPK/CDK substrates, GSK3A/B, and FOXO3 (cluster 5). In contrast, cluster 4 reflects a moderate down-regulation of 25 analytes after longer OA 100 nM treatment, such as CYP2B6, MDM2, PTEN, NFκB2, and DVL3. Early down-regulation of PAK1/2, FOXO1, and AKT1 is depicted in cluster 1 and 3, while cluster 6 shows the opposite; namely early up-regulation of PIK3R1, ERK2, HSP27, and RICTOR (Supplemental Fig. 9).
Fig. 9
Integrative analysis of protein kinase relationships. Summary of the Kinase-Substrate Enrichment Analysis (KSEA) in combination with kinase measurements by Shotgun Proteomics and DigiWest (A). KSEA (right side) results in Z-Scores where positive values indicate elevated kinase activity (orange) and negative values reduced activity (purple). The left side shows log2FC values from DigiWest (DW) and Shotgun Proteomics for matching protein kinases. Signaling network of selected protein kinases from Cytoscape using the apps OminiPath, PathLinker and Omics Visualizer (B). The inner ring shows the log2FC values measured for OA 100 nM treatment, 24 h by RNAseq and the outer ring the Z-Score from KSEA for the matching condition and kinase. Pink edges indicate stimulation in contrast to dark grey edges for inhibition
A comparison of proteins detected by Shotgun Proteomics and DigiWest yielded an overlap of 31 analytes that was explored in more detail by a heatmap (Supplemental Fig. 10). As expected, the similarity between DigiWest and Shotgun Proteomics was higher to each other than to RNAseq log2FC values. The protein RPS6 was detected in the DigiWest in the form of different phosphorylated variants and showed a clear up-regulation, while the corresponding gene was slightly down-regulated. In contrast, the bottom of the heatmap depicts a group of down-regulated proteins (e.g., DEPTOR3, MAPK3/ERK1, IDH, and RAD23B), for which transcriptomic and proteomic data are in good agreement. Remarkably, this heatmap features many genes/proteins related to the NF-κB signaling pathway, such as NFKB2, EIF4E, EIF2S1, CREB1, PDK1, and PAK2, as well multiple MAP kinases. All of them are significantly up-regulated at the transcriptomic level, while the proteomic level is not giving such a clear picture. NFKB2, EIF4E (S209), and CREB1 (S133) are increased after 24 h and MAPK1/ERK2 (T202/Y204) is increased already after 0.5 h. However, the (phospho-)protein levels of the other molecules are unchanged upon treatment or in some cases decreased after 0.5 h, e.g., PAK1/2 (S144/S141) and EIF2S1 (S51).
Integrative phosphorylation analysisTo identify which kinases might be responsible for downstream phosphorylation changes, we applied Kinase-Substrate Enrichment Analysis (KSEA). The top overrepresented kinases were PAK1, CAMK2A, AURKB, and CDK1 based on the Z-Score (Supplemental Table 4). Subsequently, Shotgun Proteomics and DigiWest data were matched to KSEA results and summarized in a heatmap (Fig. 9A) of log2FC values and corresponding Z-Scores. Most strikingly, PAK1 (S144/S141) accumulated over time as measured by DigiWest, which is reflected by an increasing Z-Score. In contrast, KSEA revealed decreased activity of MAPK3 (ERK1), which is in line with the down-regulation upon OA treatment according to DigiWest and Shotgun Proteomics. Furthermore, DigiWest data point to a slight decrease of GSK3A and GSK3B protein levels, while the phosphorylated forms of those kinases (GSK3A-S21, GSK3B-S9) accumulated over time, especially at 100 nM OA. KSEA hints to an early inhibition of GSK3A and GSK3B but slight activation after 24 h.
RNAseq results were integrated with Z-Scores of KSEA in a knowledge-based signaling network for OA 100 nM treatment (Fig. 9B). Overall, this network includes many kinases involved in MAPK, Wnt, or NF-κB signaling, such as MAPK1, MAPK8, PRKCA, PRKCG, CNSK1A1, GSK3A, GSK3B, IKBKB, and CHUK (= IKKA) as well as cytoskeleton organization similar to the network in Fig. 7B. MAPK3 (= ERK1) and PRKG2 were down-regulated at the transcriptomic level and KSEA also points to an inactivation, while PRKCD and CHUK (= IKKA) were up-regulated and KSEA hints to the same direction. However, there are a number of kinases, such as CK1, PAK1, and MARK1, for which gene expression and predicted kinase activity are somewhat contradictory, probably due to other regulatory mechanisms which are opposing the correlation between mRNA and protein levels.
Comments (0)