The PK, PD and efficacy of SB12 and ECU were studied in a total of 240 healthy subjects treated with a single dose of 300-mg intravenous infusion, and 49 patients with PNH were given recommended dosing regimen of an initial dose of 600 mg weekly for the first 4 weeks followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter.
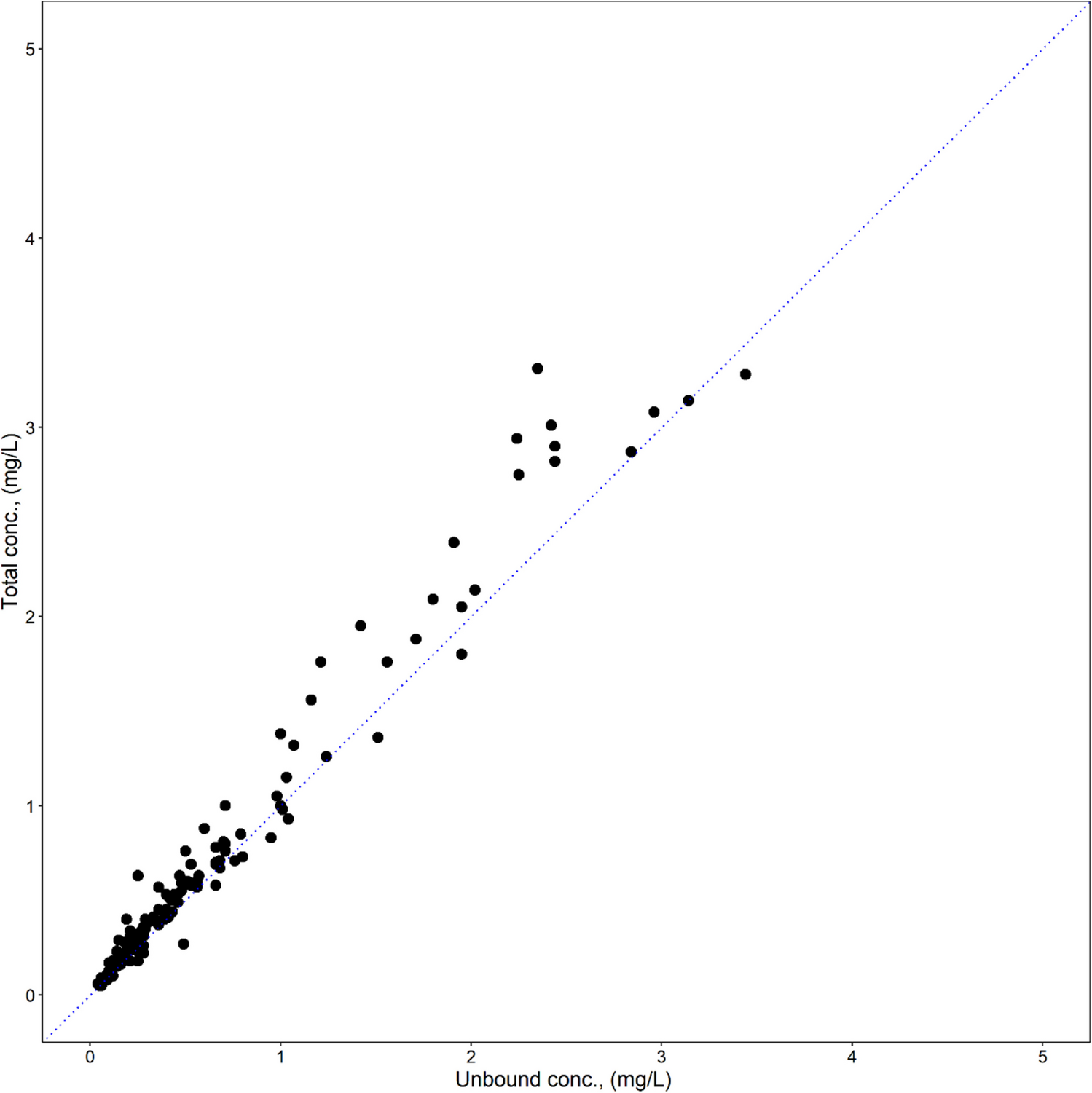
The PK observations of SB12 and ECU from phase I and phase III studies were comparable to those observed in ECU from the published references, which enabled this modeling of pooled studies with phase I and phase III data without any correction or adjustments to the serum concentrations [10, 33]. Current data from two studies had a limitation in adequately capturing the difference in PKs between healthy subjects and PNH patients. The direct comparison between healthy subjects and PNH patients was not feasible because the condition for measuring drug concentration in blood such as dosing regimen, blood sampling points and achievement status of steady-state is very different. Therefore, it was necessary to use published references for supporting the differences of PKs between healthy subjects and PNH patients [10, 33]. The differences in pharmacokinetics between healthy subjects and PNH patients were reflected in the base PK model prior to the proper covariate analysis. A two-compartment model with a proportional residual model successfully described the observed PK data, and covariate analysis identified the significant effect of weight on CL and Vc, which was in line with previous findings of ECU [10].
The PK/PD model for terminal complement activity and PK/efficacy model for LDH (not included) were separately built to evaluate the effect of serum concentration of the drug on each PD and efficacy markers separately, which may have better clinical utility and availability for the direct comparison to the reference values. In the analysis, the PD and efficacy markers were also linked in continuum to build the PK/PD/efficacy model, which allowed us to explore the relationship between terminal complement activity and LDH in patients.
The direct response model was used to link serum concentrations of the drug to terminal complement activity, which was well described by an inhibitory sigmoid Emax model. To describe LDH profiles in patients with PNH, the indirect response model inhibiting synthesis of terminal complement complex by serum concentrations was used (not included). To establish PD/efficacy models in continuum, the relationship between terminal complement activity and LDH was well described by a direct sigmoid Emax model. Other covariates potentially affecting the PD and efficacy parameters were a few laboratory parameters associated with kidney functions for terminal complement activity and red blood cells and liver functions for LDH, respectively. However, none of them were incorporated into the PD and efficacy models because physiological plausibility and evidence for the clinical significance of the identified relationships are lacking. In the PK/PD model, there was a difference between healthy subjects and PNH patients in E0 and Imax, and there was no covariate selected for efficacy model in patients. As a result of PK/PD modeling and PK/PD/efficacy modeling, there were no significant differences in PK, PD and efficacy among treatment groups in both populations of healthy subjects and patients with PNH.
In previous study, the PK/PD relationship was assessed using serum haemolytic activity in patients with RA, IMG, PNH and systemic lupus erythematous (SLE). The concentration-dependent blockade of haemolytic activity showed an inverse relationship, and higher eculizumab concentration correlated with complete inhibition of complement activity [26, 34]. Complete inhibition of complement haemolytic activity was evaluated at eculizumab concentrations as low as 29–55 μg/mL and 11–35 μg/mL in RA and SLE patients. Also, the European Medicines Agency (EMA) scientific report using pooled data from various clinical studies including single and multiple-dose studies with patients suggested that a serum eculizumab concentration of 35 μg/mL is sufficient to completely inhibit complement activity. Therefore, the dosing schedule of 600 mg in the initiation phase (i.e. weekly for the first 4 weeks) followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks was selected as the most optimal dosing regimen to achieve complete complement blockade in almost all PNH patients. The PK/PD model development in this study has some limitations. First, the effect of the relative range of sampled serum concentrations to IC50 and sigmoidicity between exposure (i.e. PK) and response (i.e. PD) on the parameter estimation performance using intensive sampling design was simulated in previous study [35]. PD parameters using sigmoid Emax model were accurately and precisely estimated when the Cmax was more than 0.85 IC50 units [35]. In case of SB12 and ECU in single-dose phase I study with healthy subjects, the Cmax value was more than 0.85 IC50 units; thus, the PD parameters estimated using inhibitory sigmoid Emax model could be judged to be accurate and precise. However, the PK/PD model using only Ctrough or steady-state data in phase III study may not precisely estimate the PD parameter, so PD parameters from phase III study may be carefully interpreted [35]. It may be related to the high inter-individual variability of efficacy parameters estimated from PD/efficacy model. Second, the IPP method was used to sequentially link PK to PD and PK/PD to efficacy for this population modeling, and the parameter estimates were relatively stable, and prediction for median value was adequate. However, there is a chance that population PK prediction and data (PPP&D) or simultaneous (SIM) method or modelling in log scale may improve the efficacy model fit [28]. Third, nonlinear elimination term (i.e. Michaelis–Menten elimination) was not applied to the PK model. At the high concentration of non-therapeutic dose range, serum ECU concentration shows nonlinear PK, and PK model integrating a nonlinear elimination allowed a better prediction of ECU concentration than a linear model [37]. Although the nonlinear elimination term was not applied because most serum ECU concentrations in this study were within the therapeutic range, two-compartment model with both linear and Michaelis–Menten elimination may more adequately describe the ECU PK.
Asian and non-Asian patients appeared to have similar SB12 and ECU PK profiles, and there were no clinically meaningful differences with regard to terminal complement activity and LDH. Also, there were no differences in PK, PD and efficacy parameters between Chinese and non-Chinese. However, it should be noted that data from Chinese subjects were only available for eight patients (16.0%) of the phase III study. Nevertheless, based on the totality of the data, it would appear that SB12 and ECU dose adjustments and dosing regimen changes are not required in specific populations (e.g. Asian and Chinese patients).
Another complement C5-inhibitor, ravulizumab, has been approved for the treatment of PNH in the US and Europe and maintains a sustained inhibition of complement for 8 weeks following intravenous infusion. Population PK/PD/efficacy modelling and simulation of ravulizumab were performed and described by the manufacturer in the approval review documents of the US Food and Drug Administration and EMA [37, 38]. The PK of ravulizumab was well described with a linear two-compartment model, with weight at baseline as covariates on clearance and volume of distribution same as ECU. This analysis confirmed that the dosing regimen of phase III is the lowest dose with maximal effect, and the dosing regimen of ravulizumab was optimized by patient’s body weight. The PK/efficacy relationship was modelled using an indirect response PK/PD model using data from patients with PNH from phase II and III studies unlike population PK/PD/efficacy modelling of ECU. The indirect response model for PK/PD relationship assumed homeostasis of the response variable prior to drug treatment governed by a zero-order production and a first-order degradation process for the efficacy variable (i.e. serum LDH). In the indirect response model, the drug-induced serum LDH lowering effect was modelled by inhibiting production of response.
One strength of this population PK/PD/efficacy modelling of ECU is the relatively large sample size of patients, particularly considering that PNH is a rare disease. Additionally, the inclusion of healthy subjects enhances the comprehensiveness of the model, resulting in a robust description of the PK, PD and efficacy profiles along with their relationships.
In conclusion, this population modeling showed PK, PD and efficacy similarities between SB12 and ECU in pooled population of healthy subjects and PNH patients, supporting the totality of evidence on biosimilarity for SB12. In addition, results of this PK/PD/efficacy analysis provide evidence-based confirmation of the recommended dosing regimen for intravenous ECU in adult patients with PNH, which comprises 600 mg weekly for the first 4 weeks followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter.


Comments (0)