
Remember me

Tuberculosis (TB) is still the leading cause of death in infectious diseases [1]. According to the World Health Organization (WHO), around 1.6 million people died of an estimated 10.6 million cases from TB in 2021, reflecting an increase of 4.5% from 2020 [2]. In addition, the COVID-19 pandemic has further compromised TB control programs [3].
Rifampicin remains a key anti-TB drug since its introduction in 1968. Rifampicin inhibits DNA-dependent RNA polymerase in Mycobacterium tuberculosis and suppresses RNA synthesis by binding to the β-subunit of the enzyme, leading to cell death. Moreover, it treats leprosy and is effective against Gram-positive cocci, including methicillin-resistant staphylococci [4,5,6].
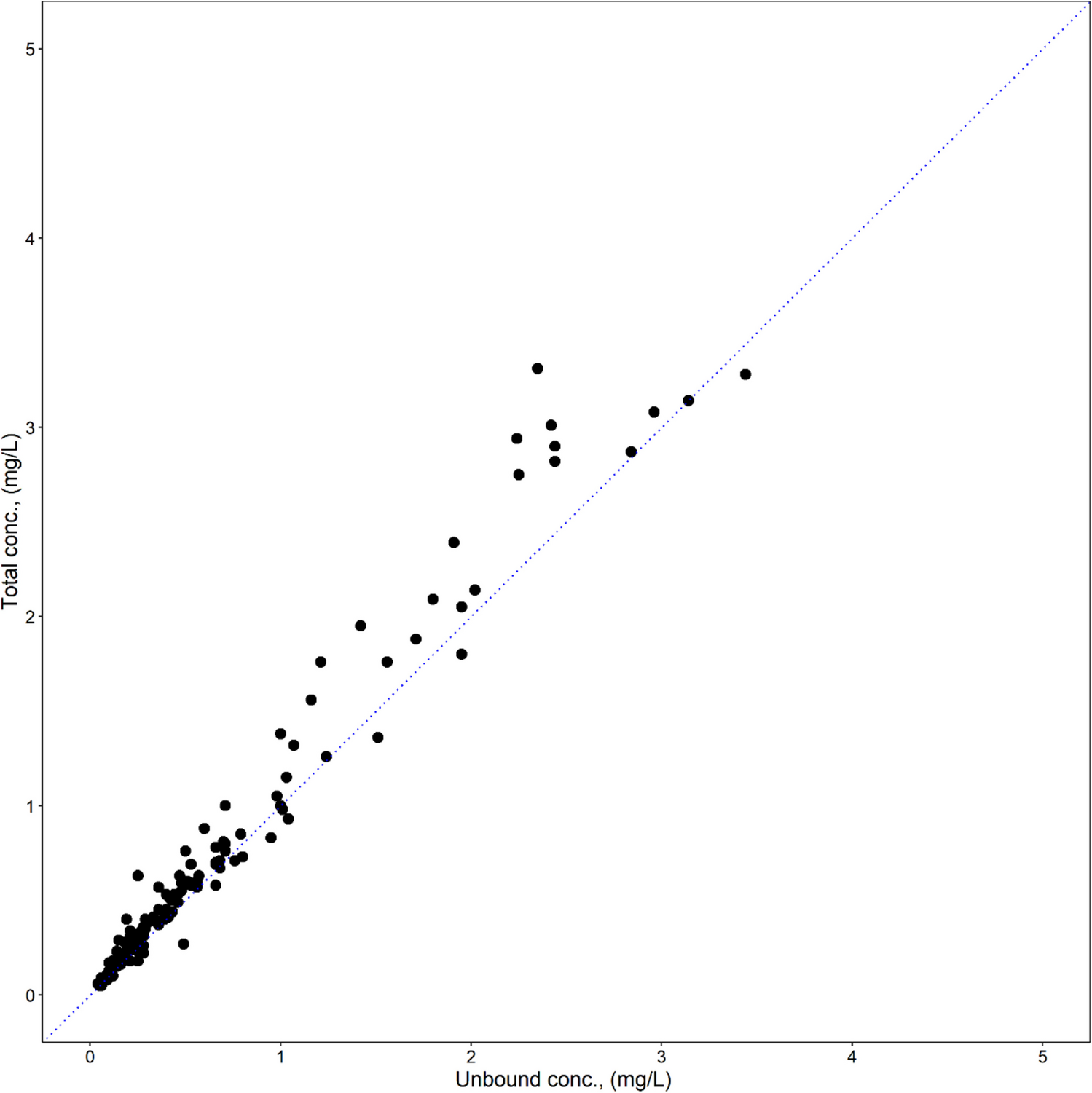
Rifampicin is readily absorbed from an empty stomach and attains maximum plasma concentrations of approximately 10 mg/L within 2 h following a single dose of 600 mg [7]. Oral absorption of rifampicin is slower when administered with food [8]. The drug is highly lipophilic, and approximately 86 to 89% is bound to plasma proteins [9, 10]. Rifampicin is quickly distributed throughout the bodily fluids, with around 5% of plasma concentrations reaching cerebrospinal fluid [1]. Plasma elimination half-life is approximately 3 to 4 h but decreases to 1 to 2 h after multiple administrations due to massive auto-induction [11]. Both rifampicin and its major metabolite, desacetylrifampicin, are primarily excreted in bile and removed in feces. Up to 30% of the administered dose is renally excreted, and only about 7% of a dose is excreted unchanged in urine [12, 13]. A greater than proportional increase in exposure in plasma is seen when the dose of rifampicin is increased (non-linear pharmacokinetics) [14]. A reduction in the exposure of concomitantly consumed medicines is frequently seen as a result of rifampicin’s extensive induction of various phase I and II metabolic enzymes and drug transporter proteins [1]. Significant induction occurs within several doses after initiating rifampicin therapy, reaches full extent in about 1 week, and disappears within about 2 weeks after discontinuation [15].
The antibacterial effect of rifampicin in patients was formerly thought to be related to Cmax/minimum inhibitory concentration (MIC), but recent preclinical investigations have shown that the area under time concentration–time curve (AUC)/MIC is better correlated with the reduction of bacterial counts [16].
It is standard practice to adjust rifampicin doses to total body weight (BW) with 10 mg/kg as the target dose [17]. Lately, fat-free body mass (FFM) was reported to be a better predictor than BW in explaining inter-individual variability of rifampicin exposure, in particular with higher doses where greater variability is expected [18, 19]. Among other possible reasons, increased hepatic metabolism related to higher body size in males was discussed to explain the higher rifampicin clearance [20]. While potential sex differences are more relevant for patients with chronic dosing, assessing such differences in healthy volunteers with a single dose and in the absence of metabolic auto-induction might help understand the background of such an effect. In the present evaluation, population (Pop) PK modeling of rifampicin was applied to data from healthy Caucasian subjects to further assess the variability of PK parameters of rifampicin and to identify the optimal body mass-related predictors of PK parameters.
MethodsSubjects and methodThe data were obtained from a phase I/IV randomized, cross-over, open-label bioequivalence study (EUDRACT-No: 2017–004418-24). The study was approved by the Ethics Committee of the Medical Faculty of the University of Cologne (18–006) and carried out in complete agreement with the pertinent version of the Declaration of Helsinki and all other relevant regulations. All volunteers provided written informed consent before participation in the study.
Study designThe study was carried out with twenty-five healthy Caucasian volunteers, with one drop-out before the first drug administration. All other volunteers completed the study, and pharmacokinetic and safety data were available in 24 individuals (11 men/13 women). Volunteers had to be between 18 and 85 years old and have a body mass index (BMI) between 18.5 and 30 kg/m2. The subjects were deemed fit for the study after extensive standard pre-study screening (medical history, physical examination, vital signs, laboratory tests, electrocardiography, etc.). Main exclusion criteria included hypersensitivity to rifampicin or any of the excipients of the preparations, any relevant clinical abnormality, smoking, chronic or acute medication, extensive ethanol consumption (> 28 g per day for males, > 14 g per day for female subjects), special dietary requirements, and history of substance addiction. Subjects had to abstain from alcohol, methylxanthine-containing beverages, orange juice, apple juice, and grapefruit products, and from extreme physical activities starting 72 h before drug administration. Pregnant and lactating women were also excluded. Participants were randomly allocated to one of the two sequences of the study, each receiving a single dose of either the test or the reference tablet of 600 mg rifampicin first and the alternate treatment after a wash-out period of at least 9 days. Test preparation was a novel rifampicin 600 mg tablet manufactured by InfectoPharm Arzneimittel und Consilium GmbH, Heppenheim, Germany, while reference preparation was a single oral dose of 600 mg tablet (EREMFAT®) manufactured by RIEMSER Pharma GmbH, Frankfurt am Main, Germany.
Blood samplingBlood samples were taken using an indwelling intravenous cannula inserted into a forearm vein. For each PK sample, up to 5 ml of blood was collected in sodium heparinized tubes at predose and 0.16, 0.33, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 3, 3.5, 4, 6, 9, 12, 16, and 24 h after drug administration. Within 30 min after withdrawal, blood samples were centrifuged at 4 °C at 1992 g for 10 min. After that, the plasma samples were stored at ≤ −70 °C until measurement.
BioanalysisThe quantification of rifampicin was carried out by using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method [21,22,23]. This process was performed by Analytical Clinical Concepts GmbH, Leidersbach, Germany, and adhered to both EMA and FDA guidelines on bioanalysis. A Shimadzu liquid chromatography system (LC-20AD Pump, Duisburg, Germany) was used for separation. The Analyst® Software version 1.6.2 (AB Sciex, Concord, Canada) was used for data acquisition, peak integration, and quantification of analytes. Rifampicin was obtained from Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany, and rifampicin (rifampicin-d8) internal standard (IS) was obtained from Alsachim, Strasbourg, France. 200 µL of plasma was mixed with 500 µL of methanol, 20 µL of ascorbic acid (0.5 mg/L), and 20 µL of the internal standard (rifampicin-d8: 100 µg/mL). After shaking the mixture at a speed of 3000 min−1, it was centrifuged at 10,500 g for 10 min (4 °C). 50 µL of the supernatant mixed with 400 µL mobile phase was transferred to a reaction vial and stored for 10 min at ≤ 20 °C. The sample was centrifuged for 10 min (4 °C) at 10,500 g, and the supernatant was transferred to an autosampler vial (HTC PAL, CTC Analytics AG, Zwingen, Switzerland). In the LC–MS/MS system, 10 µL was injected. Analytes were separated using a Kinetex® C18 chromatographic column (50 × 4.6 mm internal diameter, Phenomenex, Aschaffenburg, Germany) with a pre-column (4 × 3 mm internal diameter, Phenomenex, Aschaffenburg, Germany) and detected using an AB Sciex 2000 (Concord, Canada) mass spectrometer equipped with electrospray ionization source (TurbolonSpray®). The chromatographic separation was achieved by isocratic elution at a flow rate of 0.65 mL/min. The mobile phase consisted of 600 mL ammonium formate (2 mM), 1400 mL methanol, and 2 mL formic acid. The ion spray voltage was 4000 V, and the temperature was set to 400 °C. Ions [M + H]+ were detected in multiple reaction monitoring modes using the transitions of m/z 823.4 → 791.4 for rifampicin and 831.4 → 799.3 for IS, respectively. The column temperature was 25 °C. The linear calibration curve for rifampicin ranged between 100 and 50,000 ng/mL (r > 0.9976). The lower limit of quantification (LLOQ) was 100 ng/mL. Stability investigations during method validation showed that rifampicin was stable in plasma at room temperature for at least 6 h and during three thaw/freeze cycles (between ≤ −70 °C and room temperature). For the entire calibration range, accuracy given as a relative deviation of the mean from the nominal value was between −1.0 and 10.7%. The precision expressed in CV was ≤ 8.1% for intra-day and inter-day measurements.
Population PK analysisMonolix software version 2023R1 (Lixoft®, Antony, France) was used for non-linear mixed effect modeling [24]. The data were fitted using one and two-compartment models with linear and non-linear (Michaelis–Menten) elimination (see Fig. 1). Various absorption models were evaluated, including zero and first order, with and without lag time, and/or with transit compartments. In all models tested, elimination was assumed to take place from the central plasma compartment. The data below the limit of quantification (BQL) was defined as interval-censored at the limit of quantification, 0.1 mg/L [25]. The stochastic approximation expectation–maximization algorithm in Monolix includes simulations of the left-censored data in a right-truncated Gaussian distribution [26]. This is similar to the M4 method implemented in NONMEM to handle BQL data points [27]. Corrected Bayesian Information Criterion (BICc) was used to select non-nested models, and models with the lowest values of BICc were considered superior [28]. Inter-individual variability (IIV) was tested empirically on all PK parameters and was assumed to be log-normally distributed. The two periods were assumed to be two separate occasions, and inter-occasion variability (IOV) was tested empirically on all PK parameters. The correlation between random effects was also investigated, and a strong correlation, i.e., lowering the BICc value by more than 2 points in the non-nested models, was added to the model. To describe the residual variability, constant, proportional, and combined error models were assessed.
Fig. 1Proposed structural model. Tlag, lag time; Tk0; zero-order process;Vmax, maximum elimination rate; Km, Michaelis–Menten constant; Cp, plasma concentration
Covariate analysisIn a prior non-compartmental analysis of this study, it was confirmed that both rifampicin preparations were bioequivalent (data not shown), which allowed us to pool the data for the present analysis. Using the base population pharmacokinetic model, the potential effect of the identity of the rifampicin preparation on rifampicin PK parameters was evaluated as a covariate, along with age, sex, BW, BH (body height), body surface area (BSA), BMI, and FFM. Continuous covariates were modeled using power models normalized by weighted means, i.e., the average of the individual covariate values weighted by the number of observations per individual. Continuous covariates were modeled as shown in Eq. 1, where PKi is a PK parameter in the ith subject, PKpop is the population parameter estimation, β is the estimated coefficient of the covariate effect, COVi is the value of the covariate for subject i, and sex as a categorical covariate was modeled using a linear model where females were taken as reference. Subject characteristics used for covariate model development are given in Table 1. BSA was derived using the Mosteller formula [29], and FFM was calculated from BW and BMI for both males and females, as shown in Eqs. 2 and 3, respectively [30]. Notably, the ranges of FFM for females and males in our study population do not overlap (Table 1). Physiological plausibility and statistical significance, i.e., a reduction in objective value function (OFV) with a decrease of 3.84 (P < 0.05) for forward inclusion and an increase in the OFV of 10.8 (P < 0.01) for backward elimination [31], usual diagnostic plots (GOF plots), and visual predicted check (VPC), were the basis of selection of the final covariate model. VPC was plotted by simulating 1000 virtual subjects to compare observed data with model-based simulated data to assess the adequate predictive ability of the models. A nonparametric bootstrap analysis (1000 samples) was performed in R using the bootmlx function from Rsmlx (R speaks Monolix, version 2023.1.1) package.
Table 1 Subject characteristics used for covariate model development$$}}_=}}_}}*}}_}}i}_)}}\right)}^$$
(1)
$$\mathrm\left(}\right)=\frac^*}}^+216*}}$$
(2)
$$\mathrm\left(}\right)=\frac^*}}^+244*}}$$
(3)
Monte Carlo simulationsMonte Carlo simulations (MCS) were performed for the base model and FFM covariate model only to explore the effect of FFM on exposure to rifampicin. Using the mrgsolve package version 1.0.6 in R, 10,000 virtual subjects were simulated for a single oral dose of 600 mg rifampicin [32].
Comments (0)