
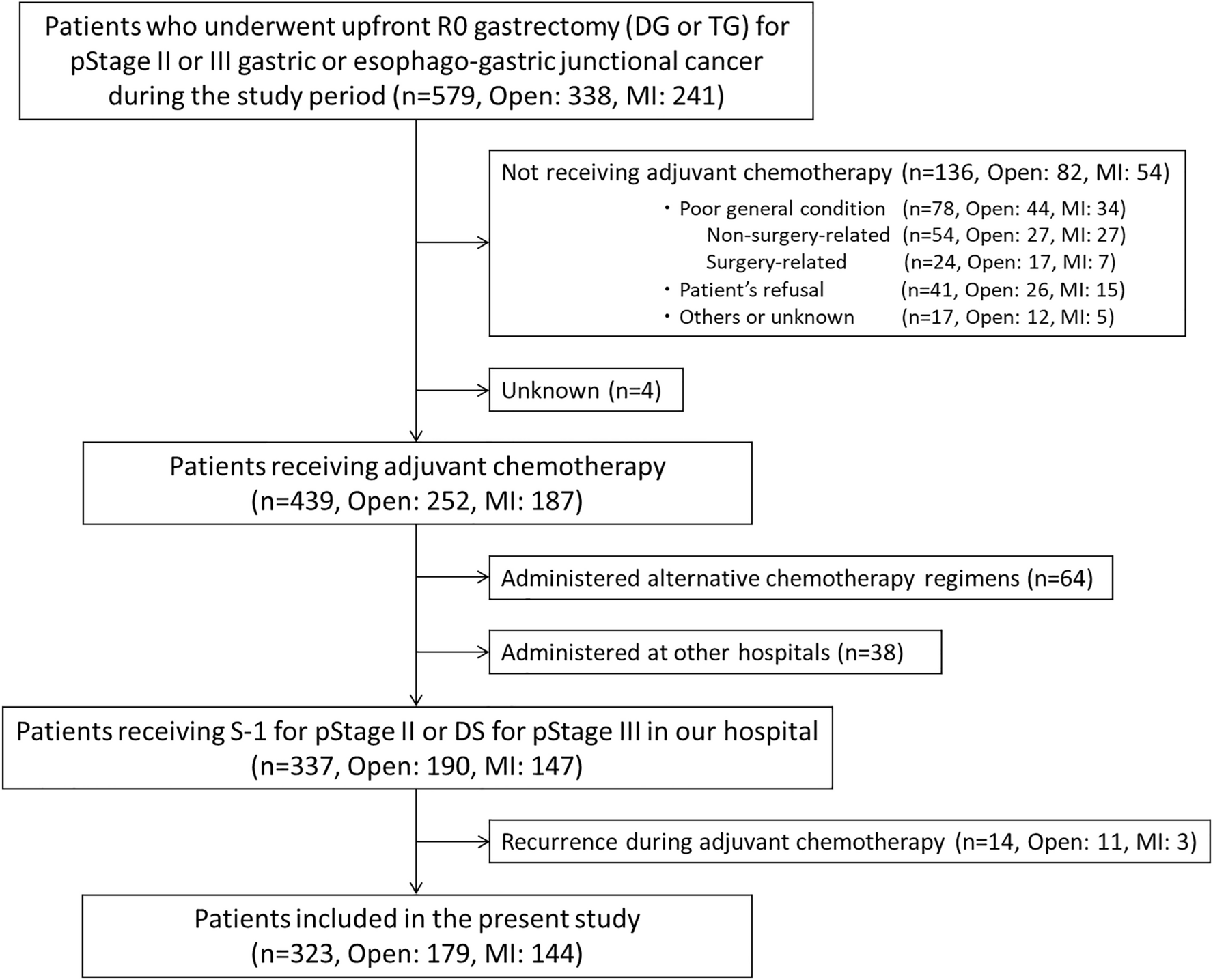
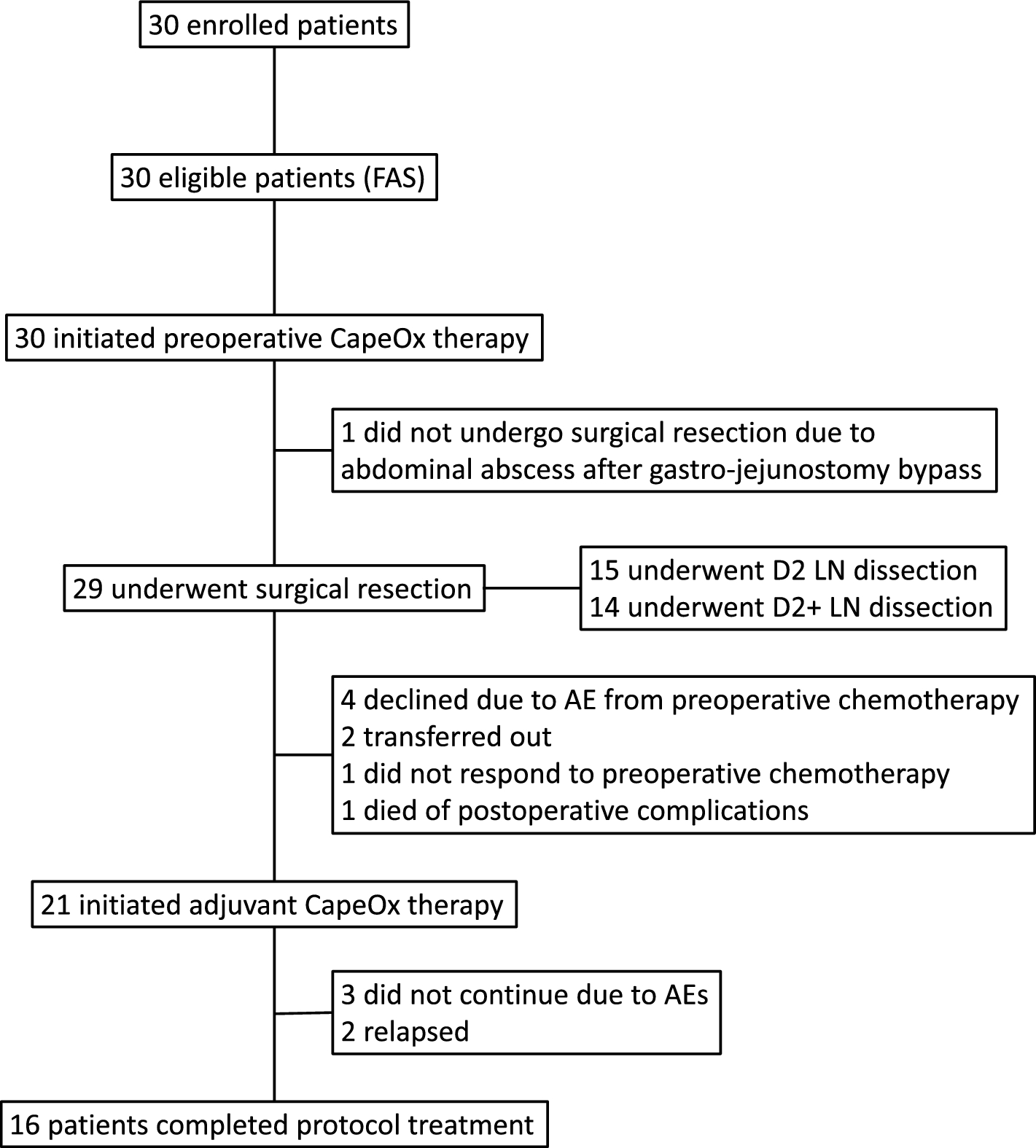
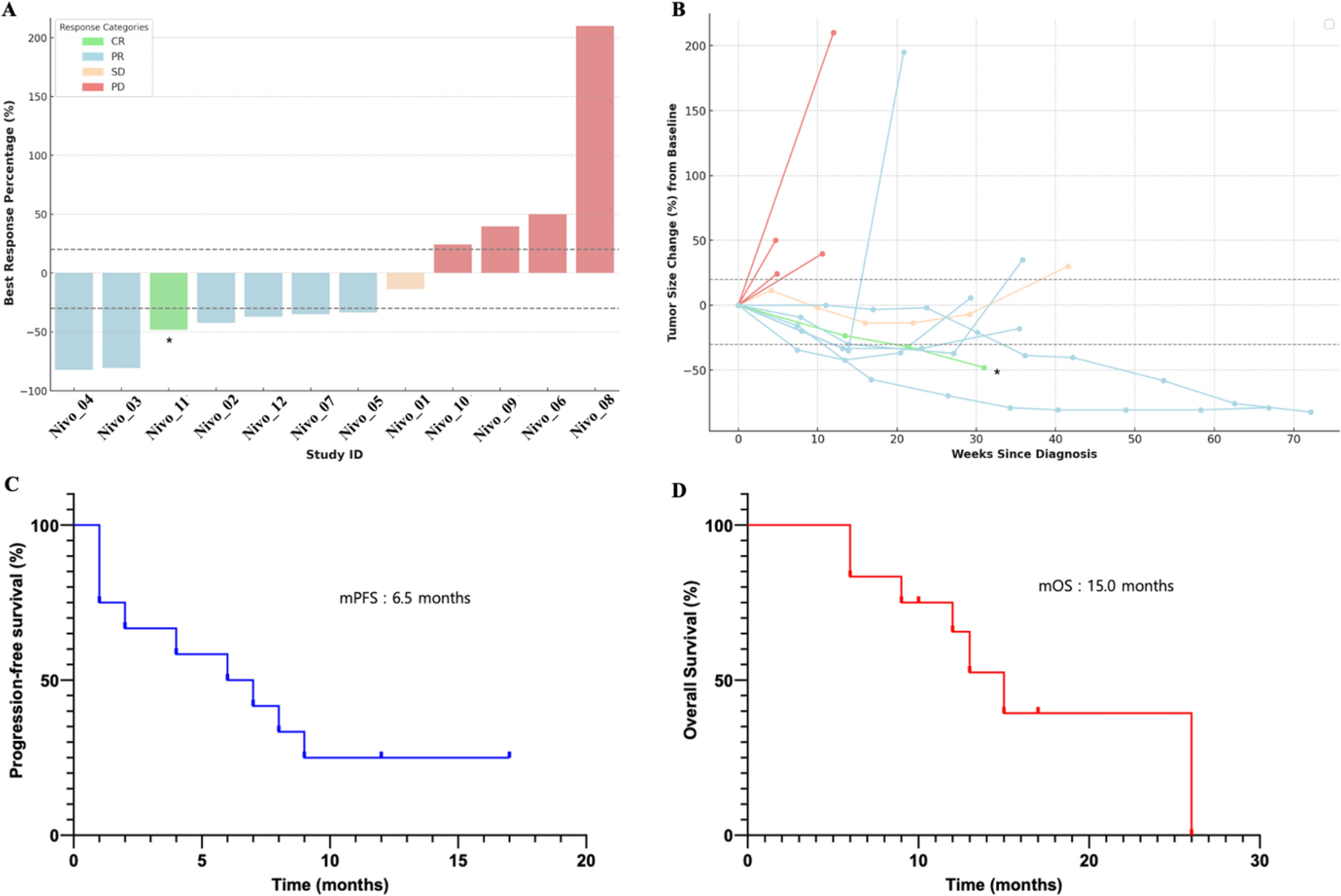
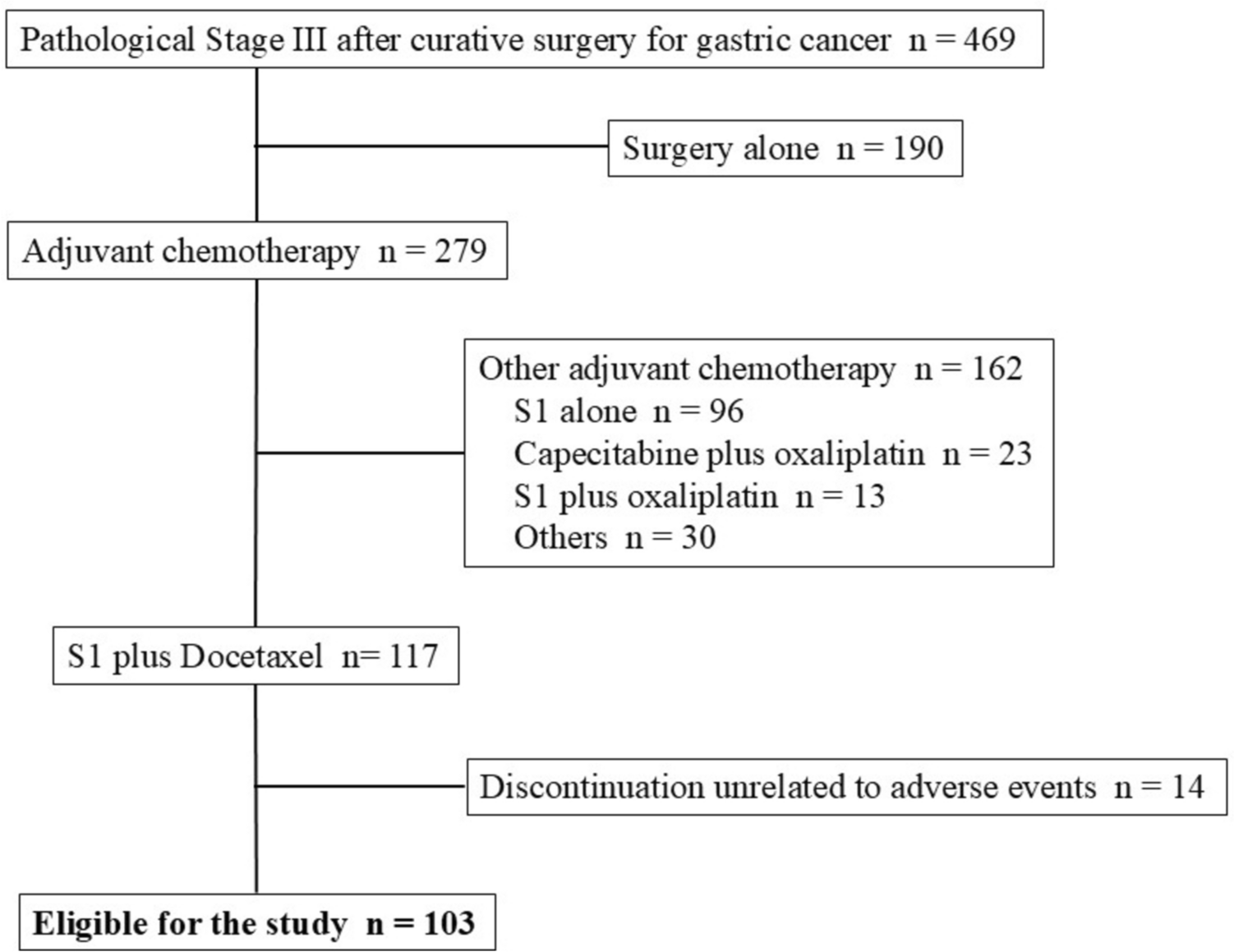
Remember me
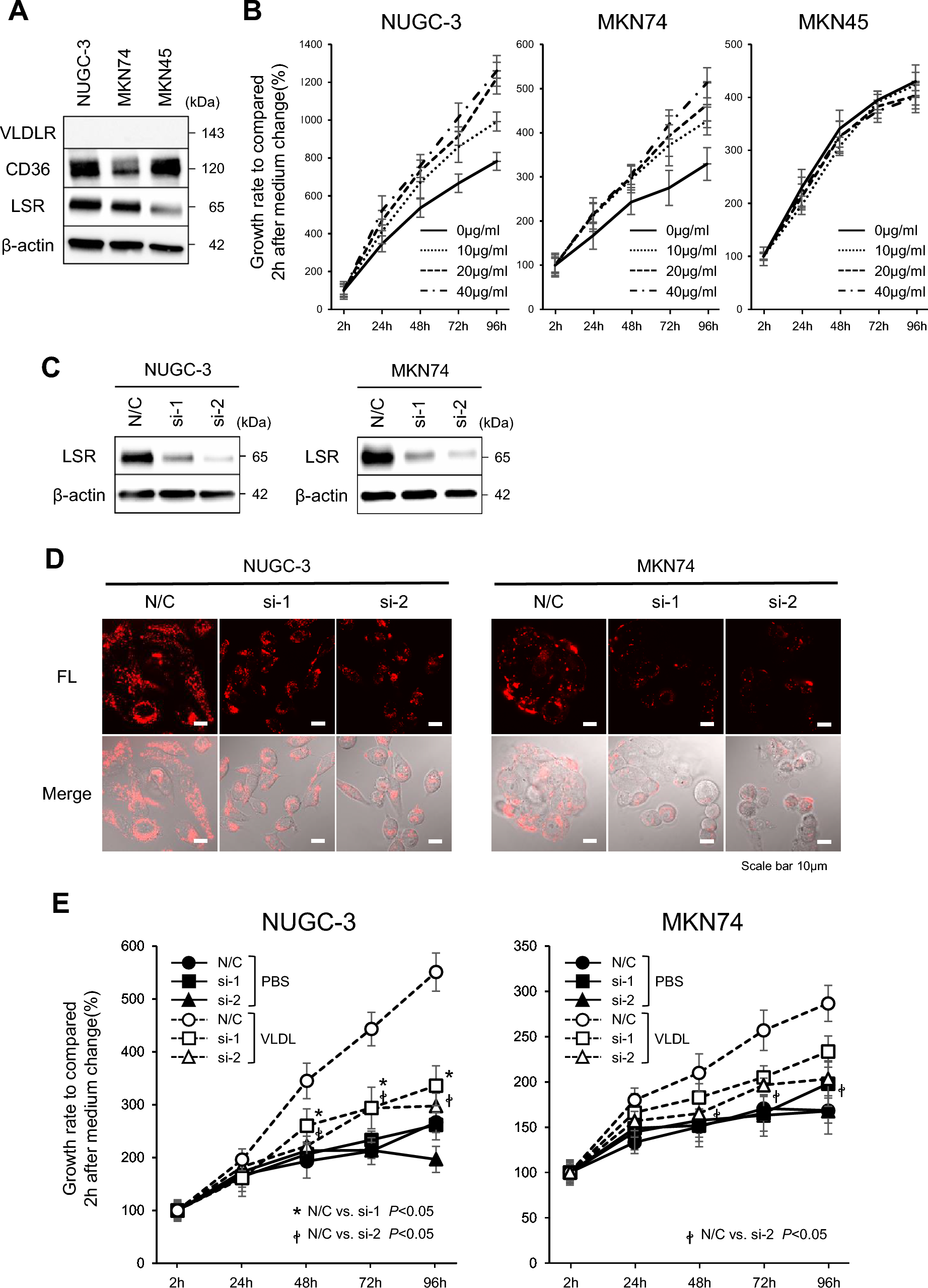
To identify molecular-targeted agent–conventional chemotherapeutic pairs with strong synergism, we combined EGFR inhibitor erlotinib, mTOR inhibitor everolimus, and c-MET inhibitor JNJ-38877605 with chemotherapeutics from five distinct groups acting via diverse mechanisms: doxorubicin (anthracycline), cisplatin (platinum derivative), 5-fluorouracil (fluoropyrimidine), paclitaxel (taxane), and SN-38 (topoisomerase I inhibitor) (Fig. 1A). We investigated the pharmacological interactions for these 15 drug pairs in AGS, SNU1, SNU5, and SNU16 gastric adenocarcinoma cell lines using Chou–Talalay’s CI method (Fig. 1B, Fig. S1A) [16]. These cell lines do not carry any loss or gain of function mutations, translocations, or fusions involving EGFR, mTOR, or c-MET. Only SNU1 cells carry a missense variation (p.T431A) in mTOR which does not affect mTOR function (Fig. 1C). The cell lines exhibited different EGFR, mTOR, and c-MET expression levels and basal phosphorylation (Fig. 1D). Hence, we tested the drug pairs in gastric cancer cells with varying target expression and phosphorylation profiles.
Fig. 1
Pharmacological interactions for the molecular-targeted agent and conventional chemotherapeutic combinations in gastric adenocarcinoma cells. A The molecular-targeted agent and conventional chemotherapeutic pairs assessed in this study. The figure was generated on BioRender. B Heatmap of combination indices (CI) at fraction affected (fa) values ranging from 0.05–0.97 for each drug pair in AGS, SNU1, SNU5, and SNU16 cells calculated by Chou–Talalay’s method. Blue indicates synergism, and white and red indicate additivity and antagonism. The strongest synergism was observed for the SN-38/erlotinib pair in SNU5 cells (highlighted by a red arrow). C The mutation status of EGFR, mTOR, and c-MET in gastric adenocarcinoma cells. D The expression and basal phosphorylation of EGFR, mTOR, and c-MET in gastric adenocarcinoma cells. E Dose–response curves for SN-38, erlotinib, and SN-38/erlotinib combination in AGS, SNU1, SNU5, SNU16, SNU484, and NCI-N87 gastric adenocarcinoma cells. F IC50 values for SN-38 in all cell lines. G CI-fa plots generated using the data in E. H Dose–response curves for SN-38/erlotinib, cisplatin–5-fluorouracil (CF), and epirubicin–cisplatin–fluorouracil (ECF) combinations in SNU5 cells. I The CIs for the combination of erlotinib with topoisomerase poisons (SN-38, topotecan, epirubicin, and doxorubicin), CF, and ECF regimens (left). The CI plots were generated using the data in E, H, and Fig. S1-C and D. Dunnett’s multiple comparisons test was used for statistical analysis. ***p < 0.001, **p = 0.001
In the pairwise pharmacological interaction screen, erlotinib and SN-38 (the active metabolites of irinotecan) emerged as the most synergistic combination (Fig. 1B). The other drug pairs exhibited antagonism, additivity, or weaker synergy compared to erlotinib/SN-38. Since these drug pairs do not provide robust models for strong synergism and are unlikely or less likely to be effective in cancer therapy, we have not pursued further investigation into the molecular mechanisms of these interactions and focused on the SN-38/erlotinib combination.
The potency of the SN-38/erlotinib combination was significantly higher than that of SN-38 and erlotinib as single agents in six different gastric adenocarcinoma cell lines (Fig. 1E). Among these cell lines, SNU5 cells, derived from the metastatic ascites fluid of a poorly differentiated gastric adenocarcinoma patient who had previously undergone chemotherapy [27], exhibited the lowest sensitivity to SN-38 (Fig. 1F). Despite that, the synergism for the SN-38/erlotinib combination was the strongest in this cell line (Fig. 1G, Figure S1B). The SN-38/erlotinib combination was much more potent than the cisplatin/5-FU (CF) and epirubicin/cisplatin/5-FU (ECF) combinations, commonly used chemotherapy regimens in gastric cancer therapy (Fig. 1H) [28].
To interrogate whether the synergistic interaction of erlotinib with SN-38 is unique to SN-38 or shared by other topoisomerase inhibitors, we tested its combination with another topoisomerase I inhibitor, topotecan, and topoisomerase II inhibitors, doxorubicin and epirubicin in SNU5 cells (Fig. 1I, Fig. S1C–D). The combination of erlotinib with all topoisomerase poisons exhibited robust synergism in contrast to the antagonistic interaction in CF and ECF. However, the degree of synergism was highest for SN-38/erlotinib combination.
Validating the synergy in SN-38/erlotinib combination dependent on cell deathThe action of drugs in cell metabolism-based screening tests may depend on partial growth inhibition and cytostasis, in addition to cytotoxicity [29]. To determine if the synergistic action of the SN-38/erlotinib combination is due to increased cell death or decreased proliferation rate, we used the FLICK assay (Fig. 2A) [30].
Fig. 2
Validating the synergistic action of erlotinib-SN-38 with the fluorescence-based and lysis-dependent inference of cell death kinetics (FLICK) assay. A The workflow for the FLICK assay adapted from Honeywell et. al. [64], created in BioRender. B Fractional viability (FV), C growth rate (GR), and D lethal fraction (LF) kinetics of the SN-38/erlotinib combination and single-agent treatments. B–D Relative doses are given in Fig. S2A. C Negative GR values indicate cell death and positive GR values indicate partial growth arrest. Untr untreated, tr treated. E Comparison of the LF curves for each treatment at relative dose 5. LF curves at all the relative doses are presented in Fig. S2C. F Quantifying LF maxima (LFmax) and area under the curve (AUC) values of data presented in E. Statistical analysis was performed using Dunnett’s multiple comparisons test. ***p < 0.001. G CI at fa: 0.5, calculated based on LF metric
The degree of cell death was evaluated using the fractional viability (FV) metric, which is simply the fraction of the total live cell population. FV analysis showed that the high potency achieved by the SN-38/erlotinib combination was due to enhanced cell death (Fig. 2B, Fig. S2A). To determine if the levels of death observed were sufficient to shrink a tumor population, we also evaluated the normalized growth rate inhibition value (GR value). Negative GR values revealed that the SN-38/erlotinib combination induced a significant shrinkage in population size, indicating high cell death (Fig. 2C). In contrast, as indicated by positive GR values, erlotinib alone caused only a partial growth arrest, whereas SN-38 led to partial growth arrest at low doses and cell death at high doses. We also evaluated the lethal fraction (LF) over time using FLICK to gain further insight into the kinetics of the death induced by these drugs. The SN-38/erlotinib combination triggered cell death more robustly than single-agent treatments (Fig. 2D–F, Fig. S2B–C). CI analysis based on the LF metric revealed a strong synergism (Fig. 2G), similar to CI values inferred from the RV-based analysis (Fig. 1E). Hence, the LF-based assessment further validated that synergism in the SN-38/erlotinib combination relied on an increased cell death rate.
To investigate growth arrest in detail, we analyzed the cell cycle progression under SN-38 alone or SN-38/erlotinib combination at relatively low and high doses. The low-dose SN-38/erlotinib combination elicited a cell cycle arrest in S-phase, also observed for high-dose SN-38 alone (Fig. 3A, Fig. S3A). However, the high-dose SN-38/erlotinib combination induced a potent cell cycle arrest in the G1 phase and increased DNA damage even at earlier time points (Fig. 3A–B, Fig. S3A–B). Next, we explored the mode of synergistic cell death by investigating the alteration in cell death kinetics in the presence of extrinsic apoptosis, apoptosis, ferroptosis, necrosis, or parthanatos inhibitors (Fig. 3C). Apoptosis inhibitor Z-VAD-FMK substantially decreased the LF under both SN-38 and the SN-38/erlotinib combination treatments, with a significantly delayed death onset (Fig. 3C–D). The incapability of the extrinsic apoptosis-specific inhibitor Z-IETD-FMK to induce a similar change indicated that the cell death by the SN-38/erlotinib combination was through intrinsic apoptosis. The assessment of cleaved-caspase3 and cleaved-PARP further confirmed prominent apoptotic cell death (Fig. 3E, Figure S3C).
Fig. 3
Phenotypic responses to SN-38 alone and SN-38/erlotinib combination in SNU5 cells. A Representative histogram for cell cycle progression (left), and percentage of cells at each phase (right), B representative flow cytometry plots (left), and fold change in p.H2AX (right) to assess alterations in DNA damage over time under SN-38 or SN-38/erlotinib combination treatment at low or high doses. PI: propidium iodide. p.H2AX: phospho-H2AX. C Identification of cell death type triggered by SN-38 alone (30 nM) and SN-38/erlotinib combination (30 nM SN-38 + 3 μM erlotinib). Lethal fraction (LF) kinetics were analyzed over time under both treatments in the presence and absence of indicated inhibitors targeting specified cell death pathways. D The death onset time, LF maxima, and AUC parameters of LF curves reporting changes in cell death rate in the presence of apoptosis inhibitor Z-VAD-FMK in C. Statistical analysis was performed using Welch’s t test. ***p < 0.001. E Analysis of apoptotic response to SN-38 alone or SN-38/erlotinib combination treatment at low and high doses via cleaved-caspase3 (c-CASP3) and cleaved-PARP (c-PARP) co-staining. Representative flow cytometry plots (left) and the kinetics of apoptotic cell death quantified (right)
The role of EGFR in synergistic actionTo investigate the dependency of synergism on the inhibition of EGFR, the primary target of erlotinib, we analyzed the fractional viability of SNU5EGFR−KO cells in response to SN-38, erlotinib, and SN-38/erlotinib combination (Fig. 4A, B), and the kinetics of cell death via LF metric (Fig. 4C). Unexpectedly, we did not observe a reversal of the synergism elicited by the SN-38/erlotinib combination in SNU5EGFR−KO cells compared to SNU5 parental cells, as FV in SNU5EGFR−KO cells was indifferent from SNU5 cells under each treatment condition (Fig. 4A–B). The evaluation of the LF also revealed that the SN-38/erlotinib combination elicited a similar cell death rate and kinetics in both SNU5EGFR−KO and SNU5 cells (Fig. 4C). These results suggested a mechanism of synergism independent of EGFR inhibition.
Fig. 4
EGFR is not involved in synergistic action. A Drug response to the SN-38/erlotinib combination and single agents in SNUEGFR−KO cells compared to SNU5 cells. Immunoblot (left) shows the lack of EGFR expression in SNUEGFR−KO cells. B Comparison of the area over the curve (AOC) and EC50 parameters deduced from fractional viability (FV) curves in A. Welch’s t test was used for statistical analysis. ns non-significant. C Dose-dependent cell death kinetics over time in SNUEGFR−KO and SNU5 cells. D Drug response to SN-38 and its dual combination with EGFR inhibitors. E Comparison of the AOC values deduced from FV curves in D, using Dunnett’s multiple comparisons test. **p value = 0.002. F Dose-dependent cell death kinetics over time under SN-38 alone and its dual combination with EGFR inhibitors. (S SN-38, E erlotinib, G gefitinib, L lapatinib, O: osimertinib, A afatinib). G CI values at fa: 0.5 deduced from LF analysis in F. H Immunoblot analysis of time-dependent alterations in ERK activity under treatment with EGFR inhibitors. I Densitometric quantitation of immunoblot data in H. The dashed box indicates the time point where a significant decrease in ERK phosphorylation was observed under osimertinib treatment but not other EGFR inhibitors, in comparison with erlotinib. Statistical significance was determined using Dunnett’s multiple comparisons test. ***p < 0.001. J Correlation between the level of ERK phosphorylation at 24 h after treatment with the specified EGFR inhibitor and CI value at fa: 0.5 for dual combinations of SN-38 with EGFR inhibitors
Several studies report different efficacies and toxicity profiles for distinct EGFR inhibitors in cancer patients [31]. To understand whether the synergistic interaction of erlotinib with SN-38 is common to different EGFR inhibitors, we tested the combinations of gefitinib, lapatinib, osimertinib, and afatinib with SN-38 in SNU5 cells (Fig. 4D–F, Fig. S4A). The dual combination of SN-38 with all EGFR inhibitors improved the drug response and enhanced the cell death rate compared to SN-38 alone, as in the SN-38/erlotinib combination, except for osimertinib. CI analysis showed that all EGFR inhibitors, but not osimertinib, exhibited synergism with SN-38 (Fig. 4G). To assess whether EGFR inhibitors’ action on other receptor tyrosine kinases (RTK) may be involved in synergism, we examined the kinetics of ERK phosphorylation, a common intracellular target of the RTK family [32] (Fig. 4H–I, Fig. S4B). Except for osimertinib, the EGFR inhibitors did not decrease ERK phosphorylation in SNU5 cells, ruling out the involvement of other RTKs as off-targets of EGFR inhibitors in synergistic response. Surprisingly, the degree of synergism with SN-38 was inversely correlated with the efficacy of EGFR inhibitors to inhibit ERK phosphorylation (Fig. 4J). Since EGFR or RTK signaling through p-ERK (phosphorylated ERK) is not altered by EGFR inhibitors that elicited synergism with SN-38, these findings strengthened that the synergism likely does not result from the loss of EGFR or RTK signaling.
Investigating the genetic dependency/vulnerability signature for drug synergy with whole-genome CRISPR screeningTo elucidate the distinct genetic dependencies underlying cell death activation via the SN-38/erlotinib combination compared to SN-38 alone, we adopted a comprehensive approach, integrating genome-wide CRISPR screening with annexin-V magnetic bead sorting (Fig. 5A). As shown in Figs. 2E and 3E, SN-38/erlotinib combination induces cell death more potently than SN-38 alone when we apply SN-38 at equimolar concentrations in both treatments. However, in equimolar concentrations, it is hard to discern whether the SN-38/erlotinib combination employs distinct mechanisms of action than SN-38 alone or if erlotinib’s presence reinforces the exact mechanisms of action triggered by SN-38 for cell death. Therefore, we identified equipotent concentrations of SN-38 and SN-38/erlotinib combination that achieve an intermediate level of cell death at a shorter drug exposure period, as opposed to previous CRISPR-based perturbation screens with DNA-damaging agents [33, 34]. Such adjustments ensured that the population size that would guarantee sgRNA representation at > 300 coverage could be maintained throughout the screen, and the sgRNA distribution of the library would mainly be affected by the alterations in cell death rate instead of the alterations in proliferation rate. Exposure to 13.5 nM SN-38 and the combination of 3.7 nM SN-38 and 370 nM erlotinib for 3 days induced ̴ %50 annexin-V positivity (Fig. 5B–D). We applied these assay conditions in our pooled screen to unveil the mechanistic traits intrinsic to cell death triggered by SN-38 and SN-38/erlotinib combination.
Fig. 5
Genome-wide CRISPR screen to explore the mechanism of synergism. A Experimental workflow of the genome-wide CRISPR screening coupled with annexin-V magnetic bead sorting. The figure was generated using BioRender. B–C Optimization of SN-38 (13.5 nM) and the SN-38/erlotinib combination (3.7 nM SN-38 + 370 nM erlotinib) concentrations and drug exposure period for CRISPR screen based on trypan-blue exclusion. Treatment with SN-38 alone and the SN-38/erlotinib at indicated concentrations for 3 days resulted in (B) fractional viability of ̴ 0.5 obtained by counting both the number of live and dead cells and (C) a ̴ 0.5-fold change in the number of live cells in the treated group compared to the untreated. D Evaluation of annexin-V positivity under SN-38 alone and the SN-38/erlotinib combination applied at optimized concentrations for 3 days. The assay conditions for both treatments achieved ̴ %50 annexin positivity. E Distribution of all genes compared to core essential genes in untreated vs. T0 sample at the gene-level zL2FC. The Kolmogorov–Smirnov test was used to calculate the p value. F Identification of candidate hit genes with an FDR < 0.1 for both SN-38 alone and the SN-38/erlotinib combination treatments, comparing the dead vs. the live populations at the gene-level zL2FC. The knockout of genes highlighted significantly altered cell death or survival rates under SN-38 alone and/or the SN-38/erlotinib. Positive zL2FC values indicate an increase in death rate, and negative zL2FC values indicate an increase in survival rate. The relationship between the two data sets was determined by computing R-squared for the genes with an FDR < 0.1. G Investigating alterations in the death rate within the knockout populations compared to the untargeted population (LacZ) under SN-38 and SN-38/erlotinib combination by analyzing LFmax. H The schematic representation of the competition assay generated using BioRender. I Results of competition assay showing the enrichment/depletion of the knockout populations compared to the untargeted population under SN-38 and SN-38/erlotinib combination. The treatments were applied at the screening doses for 3 days in (G) and (I). Dunnett’s multiple comparisons test was used to compare the knockout and untargeted populations for each treatment. ***p < 0.001, **0.002, *0.033
The quality assessment of the screen data revealed a significant decrease in core essential genes [35] in the untreated vs. T0 sample and a strong correlation between replicates (Fig. 5E, Fig. S5A). We followed a CRISPR death screen approach [36], using the comparison of “dead vs. live” at the gene-level z-scored log2 fold change (zL2FC) for the SN-38/erlotinib combination compared to SN-38 to identify differentially enriched or depleted genes under either of these treatments and both (Fig. 5F). The gene-level knockouts significantly enriched/depleted with an FDR < 0.1 in the SN-38/erlotinib combination showed a strong correlation with those in SN-38 alone. The evaluation of the LF metric under SN-38 alone and the SN-38/erlotinib combination demonstrated that deleting KEAP1 or SLFN11 decreased the death rate. In contrast, deleting ARID1B or DDX39B enhanced the death rate compared to the untargeted population, validating the screen results (Fig. 5G).
We performed competition assay in knockout populations to provide insight into the altered cell survival in response to treatments. We observed the enrichment of KEAP1 or SLFN11 knockout populations, as opposed to the depletion of ARID1B and DDX39B knockout populations in a mixed culture of SNU5-Cas9 and knockout populations under both treatments (Fig. 5H–I). However, the knockout of the genes that altered cell death or survival rates under either SN-38 or the SN-38/erlotinib combination, but not both, failed to validate the screen data (Fig. S5C–D). The high correlation between experiment groups treated with SN-38 alone and the SN-38/erlotinib combination and validation experiments suggested remarkably similar genetic dependency/vulnerability signatures for these treatments, indicating shared mechanisms of action.
Dissecting the mechanism of synergism with RNAi-based signature assayTo validate whether the SN-38/erlotinib combination employs the exact mechanisms of action with SN-38 alone, we conducted an RNAi-based signature assay, as it provides both statistical and biological generalization for the actions of anti-cancer agents [5, 7, 22]. For this, we generated the signatures of the SN-38/erlotinib combination and each single-agent treatment by assembling resistance index (RI) values for each cell population expressing one of the eight shRNAs. Then we compared the signatures to a reference set using the modified K-nearest neighbors algorithm (Fig. 6A, Fig. S6A). Our results demonstrated that the SN-38/erlotinib combination, like SN-38, clustered in the TOP1 poison category, with a linkage ratio of 1 and p value < 0.05 (Fig. 6B, Fig. S6B–C). Thus, this analysis further confirmed that the SN-38/erlotinib combination operated through the exact mechanisms of action of SN-38, as indicated by both biological and statistical classifications.
Fig. 6
Investigating the mechanism of synergism with the shRNA-based signature assay. A Illustration of RNAi-based signature assay, adapted from Jiang et al. [7], using BioRender. LD80-90: lethal dose inducing 80–90% cell death. B The heatmap generated by assembling resistance index (RI) values for each cell population expressing the specified shRNA shows the signatures of the single agents and SN-38/erlotinib combination (top). Each drug signature was compared with the reference set using the modified K-nearest neighbors algorithm that reports the linkage ratio (LR) and p value (bottom). C Principal component analysis of TOP1 and TOP2A poisons, EGFR inhibitors, spindle stabilizers, and destabilizers. Boxes in the graph highlight the estimated spatial position of each category within the PCA. The percent variance explained by each principal component and principal component 1 loadings showing each shRNA’s contribution to the analysis is provided in the table. D Drug response analysis of the SN-38/erlotinib combination and single agents in Eμ-Myc Cdkn2aArf−/− cells (left), and the calculation of CI at fa:0.5 for the SN-38/erlotinib combination (right)
We performed principal component analysis to examine the variance in our data in fewer dimensions (Fig. 6C), showing the separation of EGFR inhibitors and TOP1 poisons along the first principal component (PC1). Among the original variables from which PC1 was established, shCHK2 strongly contributed to PC1 compared to other hairpins. CHK2 is one of the crucial regulators of the signaling cascade that conveys the DNA damage signal to various downstream effectors [37]. Therefore, this data indicated that the SN-38/erlotinib combination induced DNA damage response, like other members of the TOP1 and TOP2A poisons categories, supporting our previous data in Fig. 3B. We also confirmed a strong synergism between SN-38 and erlotinib in Eμ-Myc Cdkn2aArf−/− leukemia cells, used as a cell model in the signature assay (Fig. 6D, Fig. S6E–F).
Exploring the off-target effects of erlotinibGiven that EGFR or other RTKs are not involved in the synergistic interaction of erlotinib with SN-38, and the SN-38/erlotinib combination exhibited the same genetic dependency signature as SN-38, we reviewed the literature for other targets of EGFR inhibitors that can potentiate the action of SN-38. Previous studies reported several efflux pumps as off-targets of EGFR inhibitors [38]; hence, we reassessed our genome-wide screening data to find whether the knockout of any efflux pump altered the cell death or survival rate under SN-38 only and combination treatments. We observed that the genetic ablation of ABCG2, also known as breast cancer resistance protein (BCRP) [39], enhanced the cell death rate in the screen under both treatments (Fig. 7A).
Fig. 7
The erlotinib’s off-target effect on ABCG2 is responsible for synergism with SN-38. A The screen result shows an enhanced cell death rate under SN-38 and the SN-38/erlotinib combination with the knockout of ABCG2. B Analysis of cell death kinetics via lethal fraction (LF) and C LFmax plots derived from (B). Immunoblot in B (left) shows decreased ABCG2 expression in the SNU5ABCG2−KO bulk population compared to SNU5LacZ. D Annexin V positivity (left) and survival ability assessed by competition assay (right) in SNU5ABCG2−KO compared to SNU5LacZ cells under SN-38 alone and the SN-38/erlotinib combination. E Evaluating drug response to SN-38 alone and the SN-38/erlotinib combination in SNU5ABCG2−KO and SNU5LacZ cells using fractional viability (FV). F–G Dual combination of SN-38 with ABGC2 inhibitors (ABCG2i-1 and ABCG2i-2) or with erlotinib. F Drug response analysis using FV metric (left) and the comparison of AOC values deduced from FV curves (right) in SNU5 cells. G Dose-dependent cell death kinetics over time. A1: ABCG2i-1, A2: ABCG2i-2. H FV in response to the SN-38/verapamil combination (left), comparison of AOC values (right). S SN-38, E erlotinib, V verapamil. I–J Assessment of intracellular Hoechst-33342 accumulation in a dose- and time-dependent manner under erlotinib, osimertinib, or ABCG2i-1 treatments compared to the control. I Representative flow plots (1 h). J Dose-dependent Hoechst-33342 negativity at the end of the 16-h efflux period (left) and IC50 values (right). K Relative ABCG2 expression in six gastric cancer cell lines (left) and its correlation with CI at fa: 0.5 values for the SN-38/erlotinib combination (right). Welch’s t test was used for all the panels with statistical testing. ***p value < 0.001. ns non-significant
By adopting different validation approaches, we demonstrated that knocking out the ABCG2 gene enhanced the drug-induced cell death and impaired the survival ability under SN-38 and the SN-38/erlotinib combination, which confirmed the screen finding (Fig. 7B-D, Figure S7A). Moreover, SN-38-only treatment in SNU5ABCG2−KO cells phenocopied the drug response to the SN-38/erlotinib combination in SNU5LacZ cells (Fig. 7E). Next, we investigated the drug response to the dual combination of SN-38 with different ABCG2 inhibitors, ABCG2i-1 (KO143) and ABCG2i-2 (KS176), in SNU5 cells. The combination of SN-38 with either ABCG2i-1 or ABCG2i-2 (Fig. 7F–G, Fig. S7B–C) achieved a similar degree of drug response and cell death to the SN-38/erlotinib combination. However, the ABCB1 inhibitor verapamil could not increase the response to SN-38 (Fig. 7H). Hence, these results suggested that the synergism in the SN-38/erlotinib combination exhibited a specific dependence on the inhibition of ABCG2 efflux pump activity by erlotinib.
We then investigated the effect of erlotinib on the ABCG2 efflux pump activity in a dose- and time-dependent manner by analyzing the intracellular accumulation of Hoechst-33342 dye, a substrate for ABCG2 (Fig. 7I–J, Fig. S7D–E). We used ABCG2i-1 as the positive control. Since the drug response and immunoblot findings suggested osimertinib as the only EGFR inhibitor with action on RTK activity and no synergism with SN-38 in SNU5 cells (Fig. 4D-I), we employed osimertinib as a negative control treatment. Erlotinib inhibited the ABCG2 efflux activity, but its effect was not as strong as ABCG2i-1, as expected (Fig. 7I–J, Fig. S7D–E). Osimertinib also had an inhibitory effect on the efflux pump activity at high concentrations. However, nearly an 11-fold higher dose of osimertinib was required to achieve the same degree of ABCG2 inhibition as erlotinib. These findings proved that erlotinib was a potent inhibitor of the ABCG2 efflux pump in SNU5 cells, explaining why the SN-38/osimertinib pair failed to induce a strong synergism compared to the SN-38/erlotinib combination. Moreover, the degree of synergism positively correlated with the ABCG2 expression level of gastric adenocarcinoma cell lines, except for SNU16 cells (Fig. 7K).
As supported by our perturbation screen (Fig. 5F, G, and I) and previous studies, decreased SLFN11 expression is responsible for drug resistance to DNA-damaging agents [40]. Hence, this data together suggests that the breach of the correlation between ABCG2 expression and synergy by SNU16 cells can be due to the very low SLFN11 expression in SNU16 cells compared to SNU5 cells (Fig. S7F). SLFN11 is detected as one of the top predictors of response to SN-38 by the DepMap initiative [14, 15], with low SLFN11 expression being associated with a decreased sensitivity to SN-38 in 411 pan-cancer cell models (Fig. S8A–B). Among these cell lines, the ones with high SLFN11 exhibited significantly increased sensitivity to SN-38 (IC50: ~ 10–6.72 µM) compared to the ones with low SLFN11 expression (IC50: 10–5.59 µM) (Fig. S8C). Furthermore, in cells with high SLFN11 expression, low ABCG2 expression was associated with even lower IC50 values (~ 10–7.45 µM) and higher sensitivity to SN-38 (Fig. S8D), while cells with high ABCG2 expression had higher IC50 values (10–6.59 µM). Similar to the results in cell models, low SLFN11 expression was associated with poor response to irinotecan in colorectal cancer patients (Fig. S8E–F), the response rates being high in colorectal cancers with high SLFN11 and low ABCG2 expression and low in colorectal cancers with low SLFN11 and high ABCG2 expression (Fig. S8G). All these data supported our findings in gastric adenocarcinoma cells that inhibiting ABCG2 activity in cancers with high SLFN11 expression can increase the efficacy of SN-38. These findings also explain the correlation between the synergy of erlotinib/SN-38 combination with the ABCG2 expression in gastric cancer cells with high SLFN11 expression. To understand whether this correlation is also valid in other cancers, we investigated the correlation between the CI of EGFR inhibitor/SN-38 combination with ABCG2 expression in colorectal cancer cells using data reported in the literature [24, 25]. We observed that the synergy for the gefitinib/SN-38 combination in SLFN11 positive colorectal cancer cells is correlated with the ABCG2 expression (Fig. S8H). These findings suggested that the expression level of ABCG2 could serve as one of the biomarkers for predicting sensitivity to the SN-38/erlotinib combination in cancer cells that are SLFN11 positive and inherently responsive to SN-38.
Comments (0)