
Remember me







This study will be conducted in Dutch secondary and tertiary care centers. The listing of participating clinics and hospitals can be found on www.ClinicalTrials.gov, identifier NCT05704205.
Eligibility criteriaAdult patients with moderate-to-severe AE who have inadequate disease control with their current topical treatment will be included in this study. A variety of topical treatments may have been tried including topical corticosteroids, calcineurin inhibitors, coal tar preparations, and emollients with more or less specific instructions due to the limited time in most secondary care settings. They are eligible for OTT or NB-UVB + OTT as a next step.
Inclusion criteriaIn order to be eligible to participate in this study, a subject must meet all of the following criteria:
Adult (≥ 18 years of age) patient meeting the UK working party criteria for AE [17];
AE insufficiently controlled by standard topical care and therefore eligible for OTT or NB-UVB;
Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) of ≥ 2 (moderate disease);
Eczema Area and Severity Index (EASI) of ≥ 7 (moderate disease); and
Ability to provide written informed consent.
Exclusion criteriaA potential subject who meets any of the following criteria will be excluded from participation in this study:
Contra-indication for NB-UVB [18];
Genetic defects associated with photosensitivity or skin cancer;
Heavily photo-damaged skin;
History of melanoma or non-melanoma skin malignancies (in case of basal cell carcinoma patient is excluded if one has a history of > 2 basal cell carcinomas);
Use of systemic immunosuppressants/immunomodulators;
Use of medication associated with photosensitivity;
Patient is already on systemic AE therapy;
Patient is already on OTT for the past 2 months; and
NB-UVB or any systemic therapy with influence on AD in the past 9 months.
A course of prednisolone of 15 days or more is regarded as systemic AE therapy.
Informed consentSubjects will be recruited from the patient population of each participating center. Possible eligible patients will be informed verbally about the study by a trained investigator. This may be in person or via telephone/video call. The patient will receive the participant information sheet and informed consent form (ICF) digitally or in print. Patients who agree to participate will be asked for written informed consent after reviewing the abovementioned information during a reflection period of at least one hour. In order to schedule the screening activities, it is allowed that the patient gives oral consent. The oral consent must be documented in the digital patient file. At the appointment of the screening activities the patient and the investigator will sign the informed consent form together. Allocated therapy cannot start before both parties have signed the ICF.
On the consent form, participants will be asked if they agree to the usage of their data should they choose to withdraw from the trial. Participants will also be asked for permission for the research team to share data to investigators, auditors, or regulatory authorities, where relevant. No ancillary studies are planned. This trial does not involve collecting biological specimens for storage.
InterventionsWe will investigate a course of NB-UVB + OTT (home or out-patient) of 12 weeks (range of at least 8 weeks and up to 16 weeks) compared to OTT alone. In the interventional arm, NB-UVB will be given 3 times per week with daily OTT. In the control group daily OTT will be applied.
Intervention description NB-UVBBoth outpatient clinic and at-home NB-UVB will be assessed. The ultraviolet emission from phototherapy booths and panels is made possible by UVB lamps. NB‐UVB devices contain fluorescent lamps emitting UVB in the 311 nm to 313 nm range [19].
In the out-patient clinic administration of NB-UVB rays will be executed according to the manufacturer’s instructions by trained nurses. Patients undergoing at-home therapy will receive detailed instructions on the treatment procedure prior to starting NB-UVB. During the course participants will be remotely monitored via the site’s affiliated phototherapy panel supplier, following the local procedure. During administration, all patients will wear protective goggles and cover the genital area to shield the eyes and scrotum/vulva from ultraviolet rays. Administration will occur three times a week.
The patient’s initial dosing and dosage modification during phototherapy will be done according to a standardized study dose regimen (Additional file 2). During NB-UVB treatment, the researcher (or if the researcher is not a physician: the researcher together with a supervising physician) will monitor therapy response and tolerability and determine treatment duration (minimum of 8 weeks, maximum of 16 weeks).
After completing the NB-UVB course and applying 3 months of OTT (as listed below), patients are allowed to take the next step in the AE treatment algorithm if needed, being one of the currently available systemic immunomodulating therapies.
OTTAll participants will use daily OTT. This consists of detailed instructions about the disease, corticophobia and treatments by trained nurses, bathing and showering advices, adequate emollients use, avoidance of triggers (including possible and proven contact allergens), fingertip unit (FTU) explanation and a personalized scheme of topical therapy with topical corticosteroids (TCS) of different potencies, topical calcineurin inhibitors (TCI), and tar ointments, based on the recommendations on the use of topical therapy in national and international guidelines [6, 18, 20].
In the NB-UVB + OTT group, patients are allowed to apply topical therapies two hours after UV irradiation. Patients will use OTT for at least 3 months guided by standardized informational material. All patients will be instructed to keep empty tubes of their used topical treatment (excluding emollients) and to bring them to the appointments in order to analyze potency and quantities between the NB-UVB + OTT vs OTT group.
After 3 months of OTT, if needed, patients are allowed to take the next step in the AE treatment algorithm, being one of the currently available systemic immunomodulating therapies. During follow-up, patients from the OTT group are not allowed a NB-UVB course.
Rescue medicationFor patients in the NB-UVB + OTT group, if sunburn occurs during NB-UVB, rescue medication consists of a one-time topical application of a class IV corticosteroid. For all patients, in case of severe uncontrolled AE during the study that requires rescue medication, a short course of oral prednisone can be prescribed [21]. Dosage and duration of this course will be determined by the treating physician. At the investigators’ discretion, rescue medication is allowed in other situations after consulting with the coordinating investigator.
Withdrawal of individual subjectsPatients can leave the study at any time for any reason if they wish to do so without consequences for their AE care. Investigators can decide to withdraw a patient from the study for urgent medical reasons. Reasons for withdrawal will be documented by the investigators. Withdrawal of participants has been accounted for in the sample size calculation.
If a participant prematurely discontinues study treatment, the patient will be encouraged to stay in the study to allow data collection at all remaining scheduled visits until completion of the planned end-of-study visit. The treating physician or research coordinator should aim to document the date of premature discontinuation from study treatment and the agreement of teh patient with collecting data and completing the remaining scheduled visits. Patients who fail the allocated therapy will, based on shared decision-making, continue with OTT or get screened and if eligible start with systemic immunomodulating therapy. During the follow-up period, patients will be monitored for effectiveness, safety, and satisfaction with the care received and have clinical assessments per schedule.
OutcomesOutcomes and measurement instruments from the Core Outcome Set (COS) for AE clinical trials will be used [22]. The questionnaires can be found in Additional file 3.
Primary outcomePercentage of patients with EASI50 (a 50% decrease in EASI) at 3 months follow up (range EASI 0-72) [23, 24].
Secondary outcomePhysician-reported clinical signs at 1–3–6–9–12 months:
Delta EASI
Percentage of patients with EASI50
vIGA-AD (range 0–4) [25].
Time until a 25% reduction in mean EASI is achieved
Type, frequency, and quantity of topical therapies used
Time to switch to systemic therapy
Use, amount, and duration of rescue medication
Patient-reported symptoms at 1–3–6–9–12 months:
PGA (Patient Global Assessment) (range 0–4) [26].
POEM (Patient-Oriented Eczema Measure) (range 0–28) [27, 28].
NRS-11 (Numeric Rating Scale) for peak itch over the past 24 h (range 0–10) [29].
Patient-reported quality of life at 1–3–6–9–12 months:
Patient-reported disease control at 1–3–6–9–12 months:
Patient-reported satisfaction with received treatment:
Cost-effectiveness at 3 and 12 months:
EQ-5D-5L (EuroQol 5D 5L) (range 11,111–55,555) [34].
Adapted iMCQ (iMTA Medical Consumption Questionnaire) [35].
Adapted iPCQ (iMTA Productivity Cost Questionnaire) [35].
Percentage of patients who achieve Treatment Target goals for AE at 3 (EASI50, delta PGA ≥ − 1), 6–9–12 months (EASI ≤ 7.0, PGA ≤ 2) [36].
Adverse events of special interest (AEoSIs) and serious adverse events (SAEs) at 1–3–6–9–12 months
Percentage of therapy failures and dropouts and reasons at 1–3–6–9–12 months, categorized according to Medical Dictionary for Regulatory Activities (MedDRA).
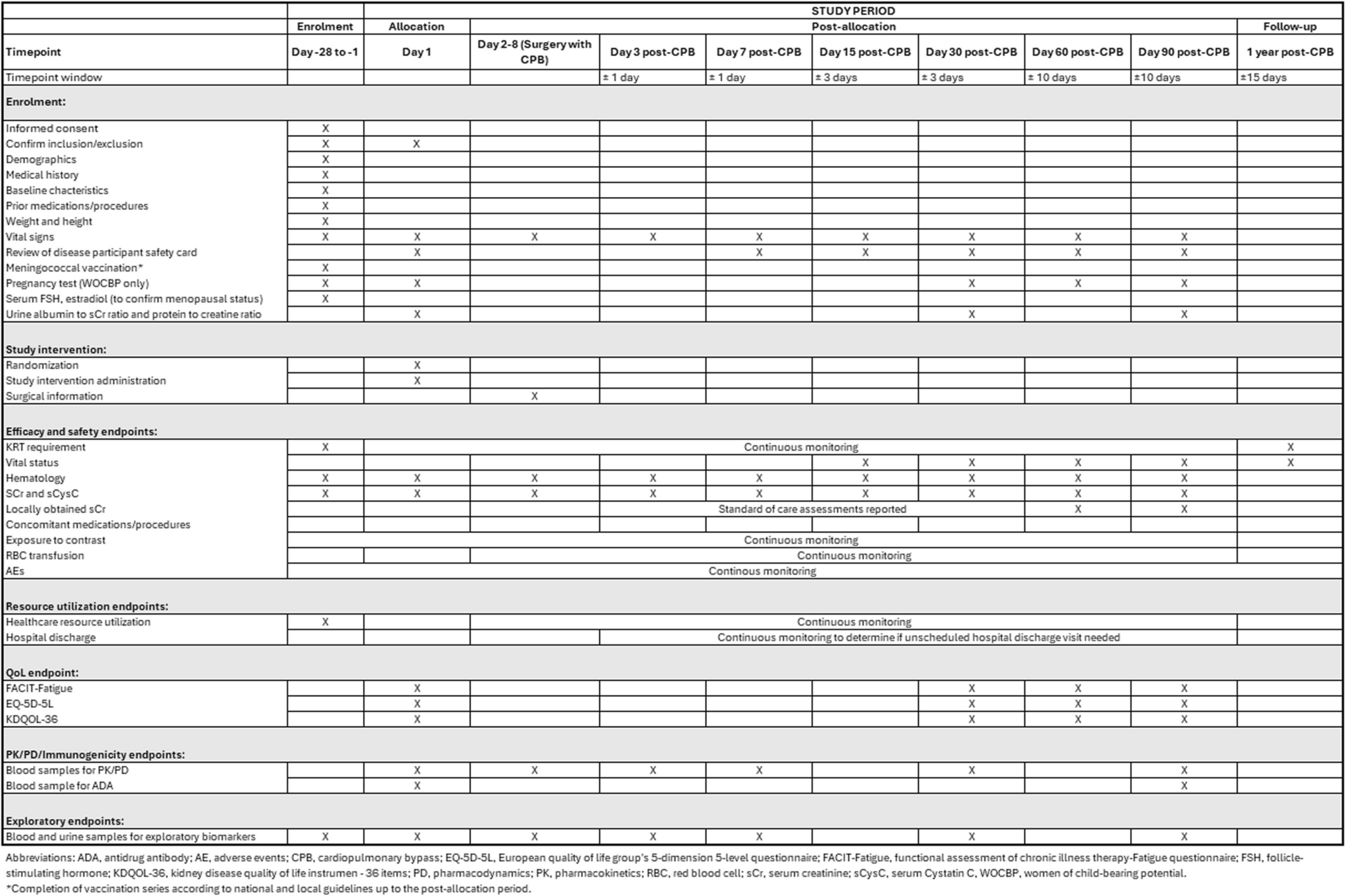
Participant timelineAfter screening and informed consent, participants will be randomized in the NB-UVB + OTT or OTT group. Study participation will entail 6 study visits. The baseline visit will take place before therapy starts. The follow-up visits are at 4 weeks, 3, 6, 9, and 12 months. Visits may be rescheduled within 1 month of the initial planning. Extra visit(s) will take place in case of therapy switch or stop. Patients in the NB-UVB + OTT group will undergo phototherapy 3 times a week for a planned period of 12 weeks from baseline, with a possible range of 8 to 16 weeks, depending on therapy response. Therapy duration will be determined in accordance to standard clinical practice. NB-UVB can be applied in the outpatient clinic or at home. Participants in both groups will apply daily OTT for a minimum of 3 months. Patients will be asked to keep all tubes of used OTT (excluding emollients) and bring them to the study visits. During the visits, data about NB-UVB and OTT will be collected. Patients will be asked for possible SAEs and AEoSIs with details such as start and stop date, intensity/severity of adverse events, probability of relation with NB-UVB + OTT or OTT, whether the allocated therapy was interrupted, and co-medications. During all visits, a physical examination by a blinded outcome assessor will take place to assess disease severity. Additionally, a set of questionnaires will be sent to the patient. The questions can be filled out on an electronic device, either at home or during the study visit in the outpatient clinic. At baseline, 3 months, and 12 months, an additional set of questionnaires regarding disease burden on productivity and medical costs will be filled out (Fig. 1). Subjects that have completed trial participation or have discontinued prematurely will be referred back to standard eczema care.
Fig. 1 Sample size
Sample sizeThe required sample size is determined for the primary endpoint EASI50 at 3 months follow-up. Based on previous research published in the literature incidences of 50% effectiveness for reaching EASI50 using NB-UVB, and of 20–30% using only OTT are to be expected [37,38,39,40].
Sample sizes of 134 in both arms achieve 90% power to detect a difference between the group proportions of 20 percentage points. The proportion in the NB-UVB + OTT treatment group is assumed to be 30% under the null hypothesis and 50% under the alternative hypothesis. The assumed proportion in the control group treated with OTT alone is 30%. The test statistic used is the two-sided Z-test pooled variance. The significance level of the test is 5%. Interim analyses for early stopping will not be conducted; hence, no correction of the significance level for interim analyses is required. Allowing for a drop-out rate of up to 15%, a total of 316 patients will be recruited.
RecruitmentSubjects will be recruited from the patient population of each participating center. Additionally, recruitment will take place via the members of the Dutch Society of Dermatology and Venereology (NVDV) and the Dutch Association for People with Constitutional Eczema (VMCE). The investigators, NVDV members, and the VMCE board will be provided with promotional material that has been reviewed by the medical research ethics committee (MREC). In addition, MREC-evaluated promotional posters and texts will be distributed via NVDV- or VMCE-based media. This includes printed advertisements in magazines and digital promotion via sites/apps such as Facebook, Instagram, and LinkedIn.
RandomizationPatients who meet eligibility criteria and who have provided written informed consent will be randomly allocated in a 1:1 ratio to either NB-UVB + OTT or OTT alone. To ensure allocation concealment, automated randomization is performed by the investigator using the electronic data management system Castor EDC. Randomization will use random permuted blocks.
BlindingDuring baseline- and follow-up visits, physician-reported outcomes (EASI, vIGA-AD) will be assessed. As these outcomes are subject to investigator bias, the physician outcome assessor will be blinded for treatment allocation. This independent (local) investigator will solely see the patient during the physical examination phase of the study visit. The patient will be instructed to not disclose their allocation outcome to this investigator. However, as NB-UVB may cause redness and tanning of the skin in light skin types, complete blinding of the outcome assessor will not be guaranteed. All other investigators or physicians performing study activities other than assessing EASI and vIGA-AD may be unblinded. The blinded outcome assessor may be unblinded when this is necessary to ensure patient safety.
Data collection and managementAll data is handled confidentially and in accordance with the European General Data Protection Regulation.
During baseline- and follow-up visits, physician-reported outcomes will be assessed by a blinded assessor who will note his/her findings on paper. This source document will be dated, signed, and kept on-site. All other study activities and the registration of physician-reported outcomes into the digital case report form (CRF) will be done by the investigator performing the study visit. Patient-reported outcomes will be entered by the participant into the electronic data management system via an invitation link, during or after every study visit. No biological specimens will be collected.
All patients will be assigned a unique subject identification number. Patients can be identified through a subject identification log and all data kept centrally will therefore be pseudonymized. This key linking patients with their identification number will be in possession of the investigators. Subject identification logs are kept on-site only and will not be shared centrally or with other sites.
Data will be collected in an electronic data management system: Castor Electronic Data Capture, from CIWIT BV, Amsterdam, the Netherlands: www.castoredc.com. Personnel responsible, e.g., data entry, monitoring, data validation, data exporting, and database administration, will have access to the database. Data handling will be described in the standard operating procedures (SOPs). All study documents will be stored securely in an environment only accessible to study team members. Data will be archived according to the local procedures of the sponsor. Data management was designed to adhere to the “FAIR Guiding Principles for scientific data management and stewardship,” aiming to optimize the Findability, Accessibility, Interoperability, and Reuse of digital assets [41].
Statistical methodsStatistical methods for primary and secondary outcomesThe primary outcome measure will be estimated as a relative risk and absolute risk difference with 95% confidence interval and p-value. The primary outcome will be analyzed using a generalized linear model with binomial distribution and log-, or identity-link respectively. The primary analysis will be run using the intention-to-treat population. Missing data for the primary outcome is expected to be low, although meaningful drop-out can be expected.
A secondary analysis will be done using the per-protocol population. Exploratory subgroup analyses will be run, e.g., location of receiving NB-UVB (out-patient clinic or at home) and below or above the median EASI score at baseline.
Dichotomous secondary outcomes will be presented as counts, percentages, and relative risks according 95% confidence interval, and p-values will be calculated using the chi-square test or Fisher exact test as appropriate. Continuous variables will be presented as means with standard deviation when data distribution is normal. In case of skewed data medians with interquartile ranges will be used for presentation. The mean or median differences between the groups will be calculated with accompanying 95% confidence intervals using Satterthwaite’s or Hodges-Lehmann’s tests. Longitudinal data (repeated measures of, e.g., EASI, POEM, NRS-11, and EQ-5D-5L) will be analyzed using generalized estimating equations and adjusted for baseline values. Time to switching to systemic therapy will be analyzed as a time-to-event outcome using a Kaplan–Meier plot and log-rank test.
Multiple imputation will be used as necessary for secondary analyses and sensitivity analyses. The effects of intercurrent events will be assessed in sensitivity analyses. A detailed statistical analysis plan will be completed before the database lock.
The economic evaluation of NB-UVB as an add-on to OTT compared to OTT alone in this study population will be performed as a cost-utility analysis (CUA) from a societal perspective with the costs per quality-adjusted life year (QALY) as a primary outcome. Health utilities will be derived from the EQ-5D-5L health status profiles using the existing scoring algorithm [42]. The scoring algorithm is based on preferences in the general population for being in a particular health state, using the time trade-off elicitation technique. The health utility score may range from − 0.44 to 1.0, reflecting the worst, respectively, best health states possible. Deceased patients will be assigned a health utility score of 0. QALYs will be calculated as the area under the curve of health utility scores over time. In addition, a cost-effectiveness analysis (CEA) will be done with the costs per patient with a EASI50 as an outcome. The analysis will include the costs of health care (in-patient and out-patient hospital care, institutional care elsewhere, out-of-hospital care like general practitioner, and extramural drugs) and costs of productivity loss resulting from sick leave from work or lowered efficiency while at work. The volume of resources used will be gathered with clinical report forms and with patient questionnaires on medical consumption (iMCQ) and productivity loss (iPCQ) adapted for this study. No interim analyses are planned.
Oversight and monitoringComposition and responsibilities of the coordinating center and trial steering committeeThis trial will be run by dermatologists, dermatological residents, (research) nurses, and PhD students in participating centers. They will be appointed, trained, and overseen by the trial steering committee consisting of the principal investigator, two project leads, a project coordinator, a methodologist, two experts in health technology assessment, a professional representative, a patient representative, and a representative of non-academic hospitals. The trial steering committee regularly meets with executive- and monitoring parties and produces progress reports to the sponsor and funders. The principal investigators, co-investigator, project coordinator, and methodologist conduct data management. All parties involved in the trial, including the participating centers, signed a consortium agreement that simultaneously serves as a clinical trial agreement. This legal document is kept in the trial master file and local investigator site files.
MonitoringMonitoring will be performed in compliance with Good Clinical Practice (GCP), the guideline of the Dutch Federation of University Medical Centers (NFU), and additional regulations, if applicable. The trial sponsor appointed Trialbureau Zorgevaluatie Nederland (www.zorgevaluatienederland.nl/trialbureaus/trialbureau) as the party that will perform monitoring. An independent qualified monitor from this party will have access to the data and source documents of the study and will perform on-site or remote quality checks on the data collection, verification of data, the rights and wellbeing of the patients, and adherence to the study protocol. All sites will be visited by the monitor at least once during the study. Central remote monitoring will occur three months after the first included patient, followed by twice yearly. All monitoring arrangements and other details regarding quality assurance will be bundled in a separate monitoring plan.
Adverse event reportingAn adverse event is an undesirable experience occurring to a subject during the study, whether or not considered related to the study activities. As this study’s investigational therapy is already standard clinical practice, we theorize that it can be accurately predicted which adverse events are study related. The known short- and long-term side effects are considered mild. Therefore we consider it safe to not include all adverse events in this study’s dataset. Investigators will instead report the adverse events that are of special interest to this study. A full list can be found in Table 1. All adverse events will be categorized according to MedDRA.
Table 1 Adverse events of special interest Adverse events of special interest (AEoSI)An adverse event of special interest is defined as an undesirable experience occurring to a subject during the study, whether or not considered related to the study activities, that is of interest to this study. All AEoSIs reported spontaneously by the subject or observed by the investigator or his staff will be recorded and analyzed (Table 1).
Serious adverse events (SAEs)A serious adverse event is defined as any untoward medical occurrence or effect that.
Results in death;
Is life-threatening (at the time of the event);
Requires hospitalization or prolongation of existing inpatients’ hospitalization;
Results in persistent or significant disability or incapacity; and
Any other important medical event that did not result in any of the outcomes listed above due to medical or surgical intervention but could have been based upon appropriate judgment by the investigator.
An elective hospital admission will not be considered as an SAE.
The investigator will report all SAEs to the sponsor (Amsterdam University Medical Centers) without undue delay after obtaining knowledge of the events. The sponsor will report the SAEs through the web portal ToetsingOnline to the accredited MREC that approved the protocol, within 7 days of first knowledge for SAEs that result in death or are life-threatening followed by a period of a maximum of 8 days to complete the initial preliminary report. All other SAEs will be reported within a period of a maximum of 15 days after the sponsor has first knowledge of the SAEs.
Follow-up of AEoSIs and SAEsAll AEoSIs and SAEs will be followed until they have abated, or until a stable situation has been reached. Depending on the event, follow-up may require additional tests or medical procedures as indicated, and/or referral to the general physician or a medical specialist. SAEs will be reported according to GCP guidelines until the study ends for the patient (when all 6 visits are completed, when the patient discontinues prematurely, or when the study is prematurely terminated).
AmendmentsAmendments are changes made to the study after a favorable opinion by the accredited MREC has been given. The MREC that gave a favorable opinion will be notified of all amendments. Should amendments to the study protocol occur, an update will be given to the clinical trial registry. Occasional deviations from the study protocol will be fully documented using a breach report form.
Dissemination plansThe investigators will adhere to the statement on publication policy made by the Central Committee on Research Involving Human Subjects (CCMO). The results of this trial will be published in peer-reviewed international medical scientific journals, preferably open access, regardless of the conclusions drawn about the investigational therapy. For knowledge sharing, (preliminary) findings will be presented at (inter)national research meetings and conferences. Participants will be informed of the results through a lay summary. This summary will additionally be disseminated to patients through the Dutch patient association VMCE.
Comments (0)