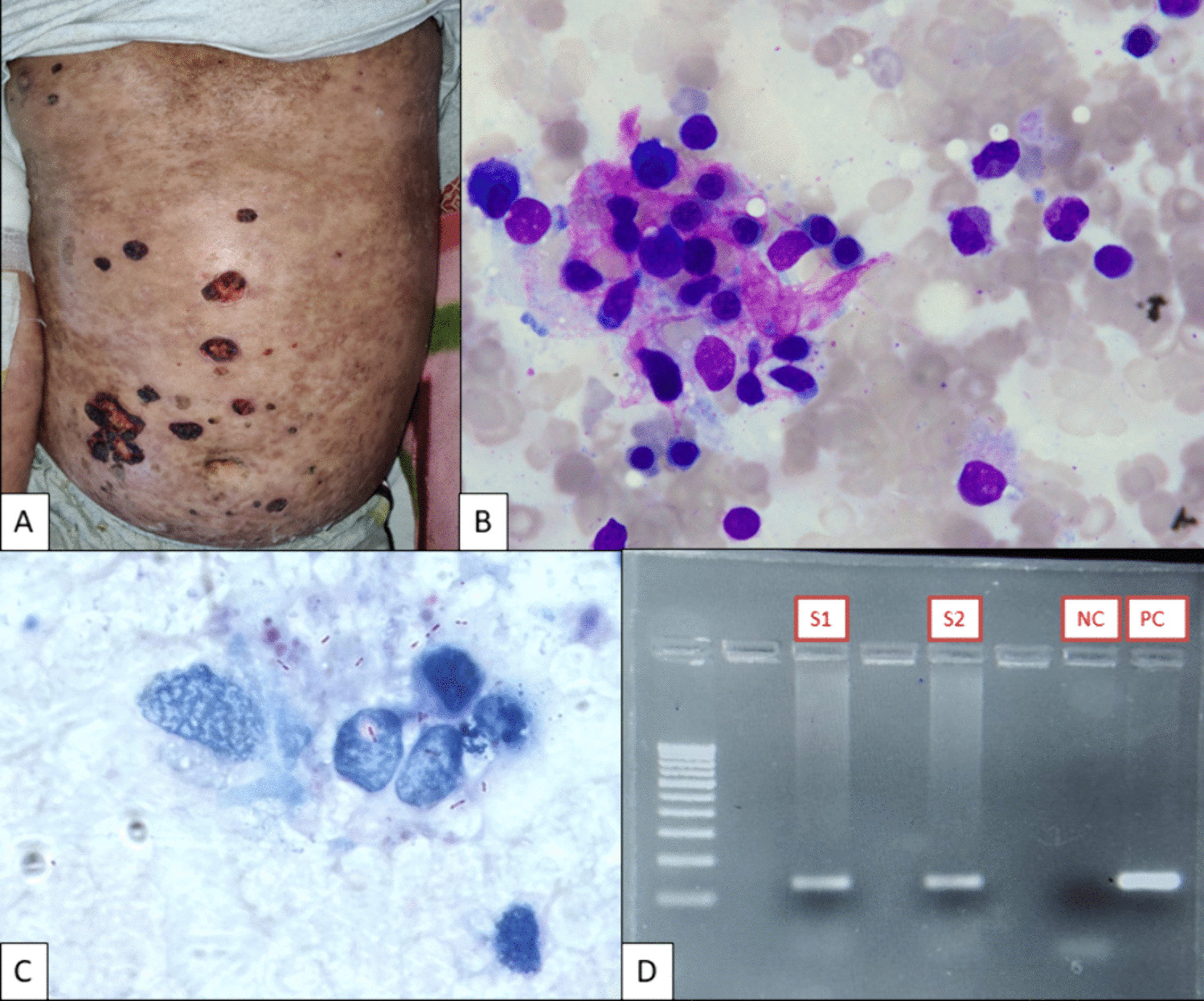
T-ALL is a T-lymphoid leukemia that accounts for nearly 15% of childhood ALL and approximately 25% adult ALL cases [4]. γδ T-ALL, in turn, accounts for only 9–12% of T-ALL [2, 3]. T-ALL is more common in adolescent males and patients typically present with a high leukocyte count, often with concurrent nodal or anterior mediastinal masses [4] The differential diagnosis in most cases includes other acute leukemias [4]. Conversely, mature γδ T-cell neoplasms include hepatosplenic T-cell lymphoma, skin and mucosal gamma delta T-cell lymphoma, and gamma-delta T-cell large granular lymphocytic leukemia (T-LGL) [4]. Microscopically, T-ALL is characterized by small to intermediate-size blasts with scant cytoplasm with occasional vacuoles, large nuclei containing condensed to dispersed chromatin, and often inconspicuous nucleoli [4]. Serial sections of lymph node in T-LBL show complete effacement of the architecture with frequent involvement of the capsule [4].
By definition, the lymphoblasts should be positive by flow cytometry for lineage-specific cytoplasmic or surface CD3 and may show variable expression of CD2, CD4, CD5, CD7, CD8, TdT, and CD1a [4]. Markers such as TdT, CD34, CD1a, CD117, and CD99 help denote the precursor nature of the blasts [4]. Aberrant expression of B-cell markers, such as CD79a, myeloid markers, such as CD13, and CD33, and NK cell markers, such as CD56, may also rarely be observed [4]. Accordingly, the immunophenotype of early T-precursor lymphoblastic leukemia (ETP-ALL) must meet all of the following diagnostic criteria: expression of cytoplasmic CD3 (with rare surface CD3 expression), absent myeloperoxidase (MPO), lack of CD1a and CD8 expression, ≥ 25% of blasts with ≥ 1 stem cell or myeloid markers (i.e., CD34, CD117, CD13, CD65, CD11b, HLA-DR), and dim-to-negative CD5 expression [4].
Cytogenetic abnormalities observed in 50–70% of T-ALL/LBL include rearrangements involving 14q11.2 (α and δ TCR loci), 7q35 (β locus), and 7p14-15 (γ locus) [4]. Other translocations include t(10;11)(p13;q14) (PICALM::MLLT10) and KMT2A (MLL) with MLLT1 (ENL at 10p13) [4]. Clonal rearrangement of TCR genes is almost always seen, and clonal IgH rearrangement may be observed in nearly 20% of cases [4]. The most common mutations affect genes involving the NOTCH1 pathway, TCR loci, epigenetic regulators (e.g., EZH2, SUZ12), chromatin modifiers (e.g., PHF6, KDM6A), JAK-STAT signaling pathway, PI3K-AKT signaling pathway, RAS-MAPK signaling pathway, and cell cycle regulators (e.g., CDKN2A, CDKN2B) [4].
To our knowledge, only a few cases of γδ T-ALL have been described in the literature, which all highlight the diverse phenotypic, cytogenetic, and molecular presentations [2, 3, 5,6,7,8]. For instance, Wang et al. and Kohla et al. describe cases of γδ T-cell neoplasms comprised of both mature and immature components; Kohla et al. also noted marked eosinophilia, which initially raised concern for a myeloproliferative process [2, 7]. Wei et al. and Fujino et al. both report cases of γδ T-ALL harboring unique AF10 fusion transcripts [3, 5] Moreover, T-ALL/LBL was formerly stratified into four immunophenotypic subtypes, corresponding to stages of normal T-cell differentiation: (1) pro-T/T-I, (2) pre-T/T-II, (3) cortical T, T-III, and medullary T/T-IV [4]. Although γδ T-cells undergo thymic maturation, they do not incorporate CD4 or CD8 co-receptors and do not function through conventional MHC binding mechanisms. Interestingly, while we observed CD4 expression in our case, all reports of γδ T-ALL to date have described dim-to-absent expression of CD4 and CD8 [2, 3, 5,6,7]. Lastly, when compared to its αβ counterpart, γδ T-ALL was previously reported to have poorer prognosis with a CD45RA−/CD45RO+ phenotype and lower hemoglobin levels in children and splenomegaly with higher white cell counts in adults, although further studies are needed to understand the distinctions [8].
Preliminary results from a comprehensive genomic analysis based on whole-genome sequencing (WGS) and RNA-sequencing data identified several genomic subtypes of γδ T-ALL, with the STAG2/LMO2 subtype representing a high-risk group (age at diagnosis < 3 years with poor survival outcomes) [9]. Previously, based on results from whole-exome sequencing, single nucleotide polymorphism (SNP) microarrays, and RNA transcriptome sequencing of children and young adults with newly diagnosed T-ALL enrolled on the COG trial AALL0434, 242 patients were classified into eight subgroups characterized by deregulation of TAL1, TAL2, TLX1, TLX3, HOXA, LMO1/2, LMO2-LYL1, or NKX2-1 [10] Although additional comprehensive genetic studies are needed, as mutations in several signaling pathways overlap in many cases [10], γδ T-ALL requires early recognition and prompt work-up to distinguish it from ETP-ALL and mature γδ T-cell neoplasms.










Comments (0)