DLBCL is an aggressive and heterogeneous grouping of non-Hodgkin lymphomas encompassing a spectrum of different immunophenotypic and molecular variants. The definition and categorization of DLBCL with multiple gene rearrangements (so-called hits) have been evolving in recent years. The 2022-ICC retains a subgrouping for cases with MYC and BCL6 rearrangement; this is recognized as a heterogeneous category with variable gene expression profiles and mutational spectra [2]. Neither the WHO nor ICC classification schema recognize HGBL with MYC, BCL2, BCL6, and CCND1 rearrangements (a so-called quadruple hit) as a unique entity.
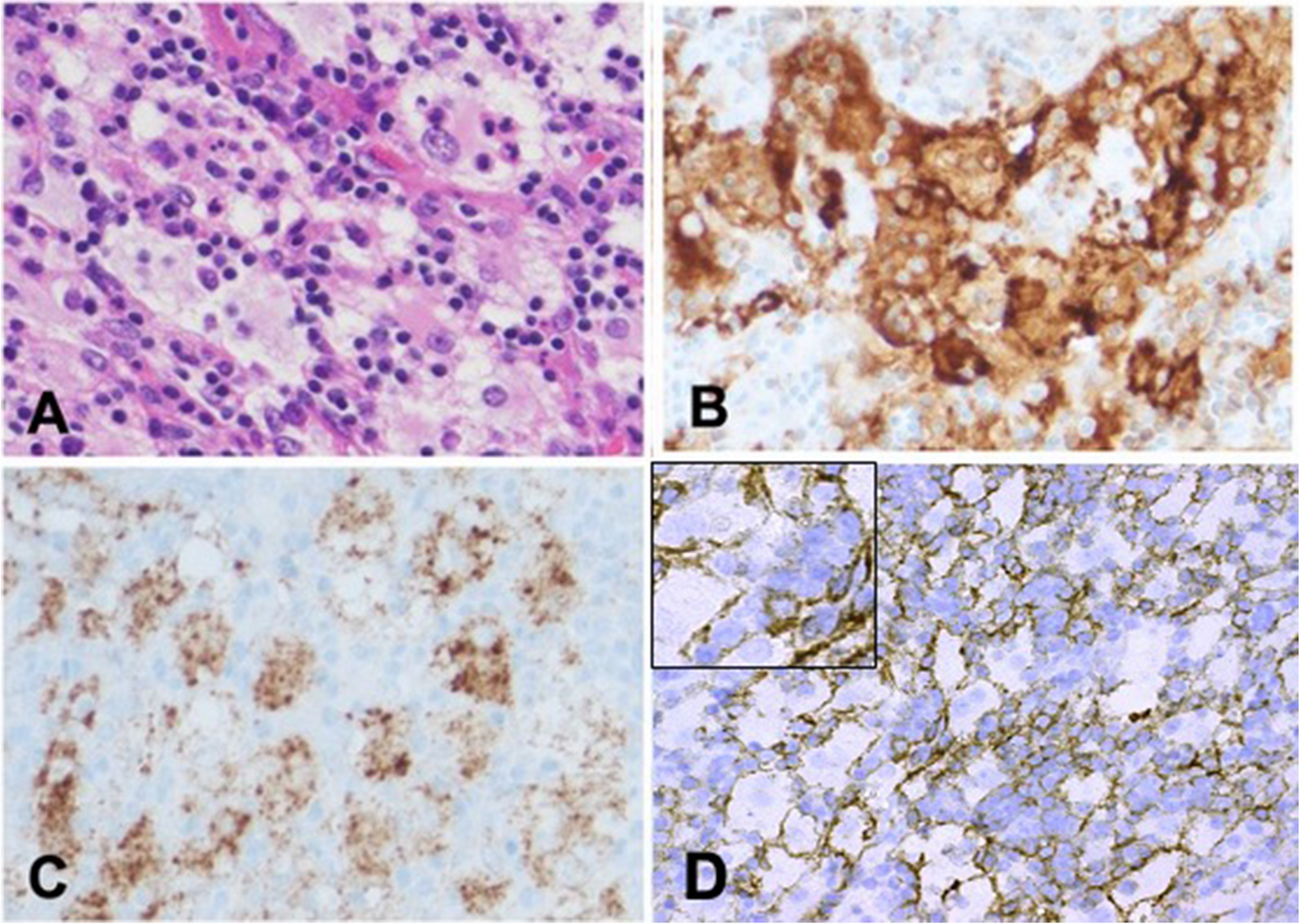
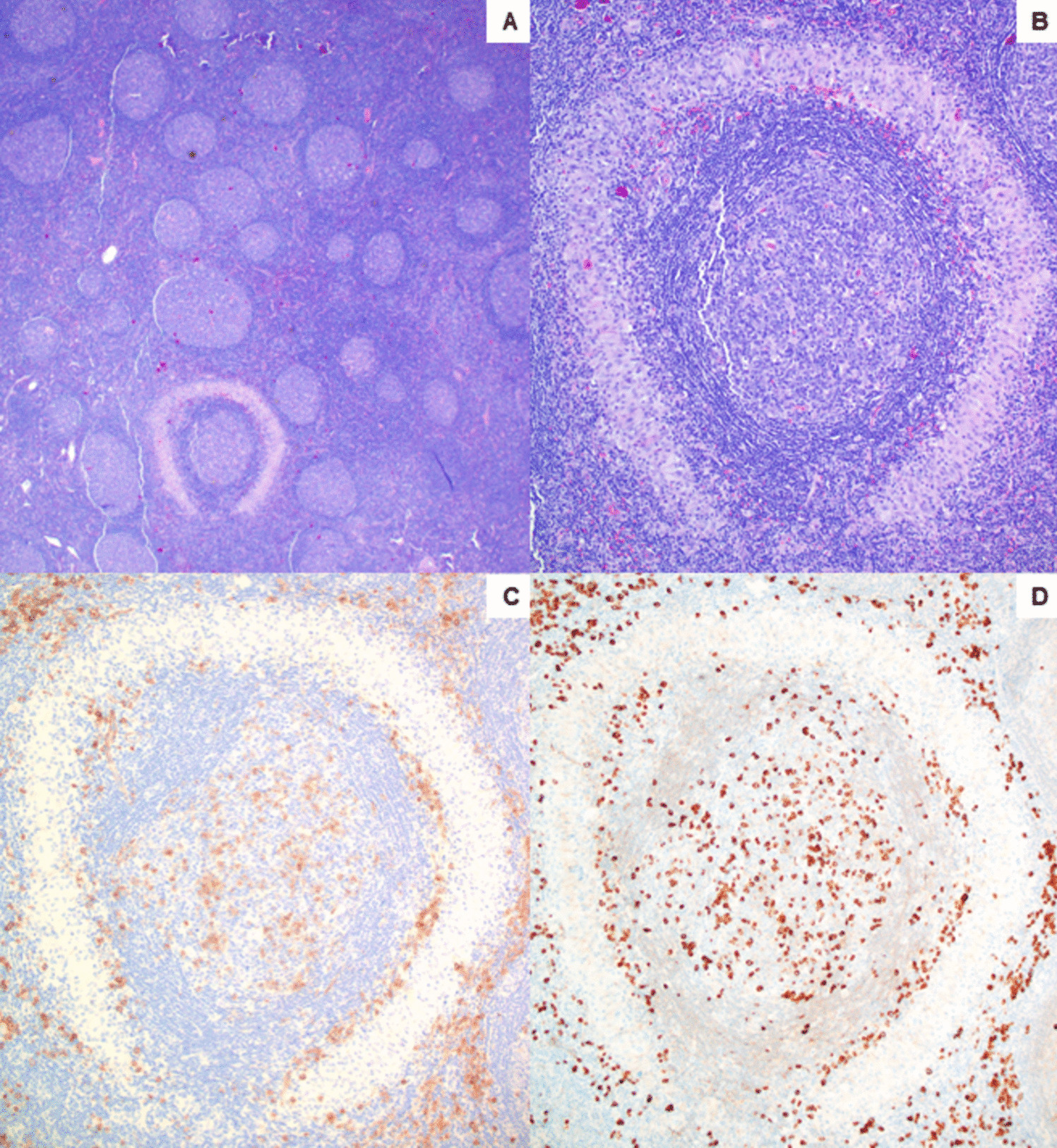
The tumor cells in our case showed mild pleomorphism with eosinophilic nucleoli and some blastoid chromatin. Tumor cells were positive for BCL1, raising the possibility of a blastoid/pleomorphic variant of mantle cell lymphoma (MCL). However, CD5 and SOX11 were negative, arguing against a diagnosis of MCL. TdT was also negative. Tumor cells were also positive for CD10 and BCL6 (subset), further supporting germinal center derivation. Following the currently accepted classification schema, our case is best classified as a HGBL with MYC and BCL2 rearrangement.
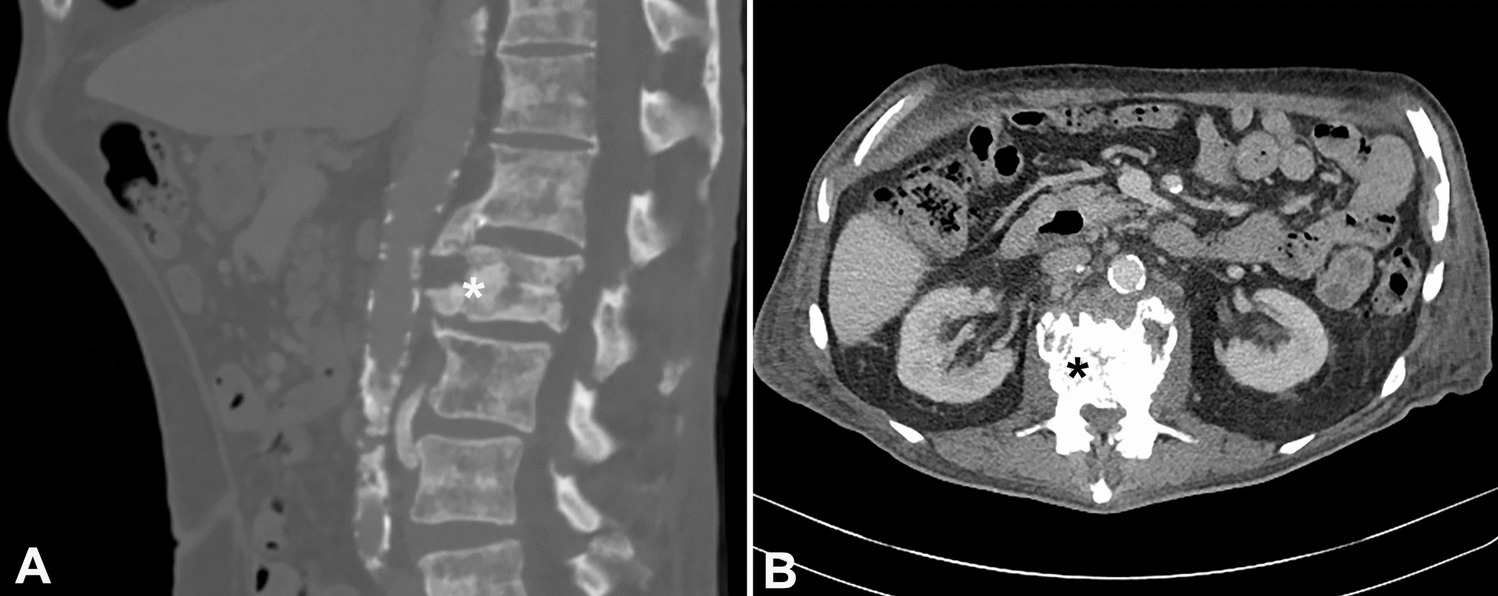
B-cell lymphomas with MYC, BCL2, BCL6, and CCND1 rearrangements are rare entities. Available outcomes data from previous publications suggest this entity has a poor prognosis [3,4,5,6,7,8,9,10]. Among the reported cases, there were four males and three females with median age 74 years (range 51–81 years); lymphadenopathy was seen in four of the seven cases. Two cases reported staging information, and both were stage III. Most cases were negative for CD5 and SOX11 with Ki-67 in the range of 60–90%. Demographic data was not provided for the three remaining cases. There is no consensus on the optimal treatment and the outcomes have been dismal, despite aggressive initial therapies [7]. The clinical features of all reported cases are summarized in Supplementary Table 1 and the histopathological features, diagnosis, ancillary studies, treatment, and outcome/overall survival are summarized in Supplementary Table 2.
CCND1 gene rearrangement or other genetic alterations involving the gene can lead to aberrant BCL1 protein expression. Among hematolymphoid tumors, BCL1 is found to be overexpressed in > 90% of MCL cases and about 40% of plasma cell myelomas, both of which are caused by a translocation which juxtaposes the immunoglobulin heavy chain (IGH) gene to the CCND1 gene. BCL1 protein expression is rarely seen in DLBCL, which has been linked to copy number gains of CCND1 or via mRNA dysregulation [11, 12]. FISH studies in our case demonstrated additional signals of CCND1, a likely mechanism for the observed protein expression by IHC.
In DLBCL, the data suggests that the CCND1 rearrangement is a secondary event during lymphoma evolution [12]. Cheng et al. included a quadruple hit lymphoma in their report of DLBCL with CCND1 rearrangements considered to represent secondary genetic events [9]. This is in contrast to MCL, where CCND1 rearrangement is considered to be a primary genetic event [12]. MCL may also gain secondary BCL2, BCL6, and MYC rearrangements, as proposed in three [3, 9, 10] of the previously reported quadruple hit lymphomas.
In our case, it is difficult to confidently determine the sequence of genetic alterations. All translocations were found in relatively (and similarly) high proportions of tumor cells. However, sequencing results were more typical of DLBCL than MCL. The combination of mutations best fits in the EZB-DLBCL molecular cluster according to Morin et al. [13]; similar reported molecular clusters include C3, BCL2, and MYC/BCL2-DH [14]. Interestingly, a previously sequenced quadruple hit lymphoma, designated as a pleomorphic MCL, shows limited overlapping mutations with our case [10]. The PIM1 gene was the sole shared mutation with our case. Mutations in the PIM1 gene are frequently seen in lymphomas and have been implicated in DLBCL pathogenesis [15]. Although PIM1 mutations identified in our case are variants of uncertain significance (VUS), three out of the four mutations (c.87G > C, c.68C > T, and c.286G > C) have been reported multiple times in DLBCL [16,17,18] respectively; the other PIM1 mutation (c.113A > T) has not been reported to our knowledge. NGS findings from the present case and the two previously sequenced quadruple hit lymphomas are summarized in Supplementary Table 3.
MYC mutations in DLBCL are more frequently seen in cases with MYC and BCL6 rearrangement, which was present in our case [19]. This is thought to be due to aberrant somatic hypermutation from activation-induced cytidine deaminase, also implicated in the genesis of MYC rearrangement [20]. All three MYC mutations in our case are identified as VUS and have not been previously reported. Cases exhibiting intact (non-rearranged) copies of MYC on break apart probe testing, including some cases with additional intact MYC signals, as in our case, have revealed the presence of MYC gene fusions when followed by an IGH::MYC dual fusion probe [21].
FISH studies also showed additional copies of BCL6. This finding has been seen in nearly half of MYC-rearranged DLBCLs according to one study [22].
KMT2D is frequently mutated in DLBCL (~ 30% of de novo cases, including both germinal center B-cell and activated B-cell subtypes and follicular lymphoma (~ 90%)) [23]. Recent studies have suggested KMT2D mutations represent early events in a common progenitor before divergent evolution of follicular lymphoma or DLBCL, the latter occurs through acquisition of additional genetic lesions and clonal expansion [23, 24]
Among lymphomas, TNFRSF14 mutation has been reported in follicular lymphoma as well as DLBCL, NOS (EZB) [25] TNFRSF14 mutations have not been reported in mantle cell lymphoma [9]. The specific TNFRSF14 mutation found in our case (c.472C > T) has been previously reported in follicular lymphoma and DLBCL [26].
In addition to BCL2 translocation, our case also harbored BCL2 gene mutation (c.585 + 4G > C). This was categorized as a VUS for our case but been previously reported in follicular lymphoma and DLBCL [27].
B-cell lymphomas with concurrent MYC, BCL2, BCL6, and CCND1 rearrangements appear to be a rare occurrence; however, current standard approaches to DLBCL/HGBL classification do not require routine testing for CCND1 rearrangement. With the current classification schemes de-emphasizing the importance of BCL6 rearrangement, this may no longer be routinely assessed as well. The addition of CCND1 rearrangement in the workup for a DLBCL/HGBL might only be sought in cases with BCL1 protein expression, as seen in our case. Sequencing may be of benefit for delineating DLBCL from MCL in the quadruple hit setting, although current data is limited.






Comments (0)