
Remember me


The INDYGO trial was a prospective, non-randomized, single-centre, open-label, phase I study (NCT 03048240). Patients were eligible if they were 18 years of age, had a newly diagnosed glioblastoma (according to the WHO 2016 classification) with an indication of possible gross total surgical resection of the contrast enhancement. All potential candidate cases were discussed in a multidisciplinary neuro-oncology meeting and inclusion was confirmed. Patients were followed every 3 months until progression or for 30 months, whichever came first.
The study was conducted in accordance with the principles of the Declaration of Helsinki. The protocol was approved by The French National Agency for Medicine and Health Product Safety (ANSM) and reviewed by the French National Ethics Committee. The study was designed in collaboration between the study sponsor and the investigators.
All patients gave written informed consent, according to institutional regulations. The data were analysed and interpreted by a sponsor biostatistician in close collaboration with the investigator. The trial management committee and academic investigator had access to the raw data.
SettingPatients were selected and enrolled at a single tertiary neurosurgical centre, the University Hospital of Lille, between May 2017 and June 2018. The last visit of the last included patient took place in April 2021. The database was locked in September 2021 and the study was closed in October 2021. After the end of the clinical trial, patients were followed as part of the standard clinical follow-up and additional survival data were collected.
Eligibility criteriaInclusion criteria for the study were as follows: age ≥ 18 years; Karnofsky Performance Status (KPS) score ≥ 60; suggestive cerebral MRI findings suggestive for a HGG; absence of contraindications to MRI; surgical indication; histological diagnosis of glioblastoma; ability to undergo the SOC after surgery; absence of contraindications to MRI; absence of contraindications to 5-ALA HCl (i.e. adequate hepatic and renal function: bilirubin < 1.5 times the upper normal value; serum aspartate aminotransferase, alanine aminotransferases and alkaline phosphatases < 2.5 times normal value; creatinine clearance > 30 mL/min).
Key exclusion criteria included: multifocal disease; hypersensitivity or contraindications of 5-ALA HCl and/or porphyrins; acute or chronic types of porphyria; use of other photosensitizing drugs, history of cardiopulmonary disease; soy allergy; pregnancy or nursing.
Study participants12 patients were enrolled, 10 patients with surgically accessible lesions for maximal resection were analysed and 2 were discontinued (one due to a liver dysfunction contraindicating the use of 5-ALA HCl and one due to the intraoperative diagnosis of a left temporal abscess).
One patient at a time was enrolled in the study to allow prospective analysis of safety during and immediately after the procedure. Following the inclusion of the first 5 patients, an independent safety monitoring board (ISMB) met and approved the completion of the trial with the inclusion of five additional patients.
Baseline patient characteristicsBaseline patient characteristics are described in Table 1. The median age was 57.1 years [35–69.3], 70% of the patients were male, and the median KPS score was 85 (range 70–100). All patients had a histologically confirmed glioblastoma with IDH mutation present in only one patient (#10). Four patients presented with a hypermethylation of the MGMT promoter gene (#04, #06, #09 and #10).
Table 1 Baseline characteristicsIntraoperative PDT descriptionIntraoperative PDT procedure (Fig. 1) has been described in a previous article [25]. It consisted of the orally delivery of Gliolan® as photosensitizing drug, a fluorescence-guided gross total resection assessed by intraoperative MRI. Additional resection could then be performed, followed by the illumination of the tumour bed with a specific prototype illumination device. An immediate postoperative MRI was performed early after intraoperative PDT to confirm the absence of an acute adverse event.
Fig. 1
Workflow of the intraoperative PDT procedure; iMRI: intraoperative magnetic resonance imaging, FGR: fluorescence guided resection, PpIX: protoporphyrin IX
The illumination device is composed of a red wave-length light diffuser as illustrated in Fig. 2. The distal balloon is inserted and inflated with a intralipid diffusing solution to fit the shape of the resection cavity [27]. Light is emitted through an optical fiber connected to a medical non thermal laser device and diffused within the balloon.
Fig. 2
View of the light diffuser (A), theoretical (B) and intraoperative methodology (C) for insertion in the surgical cavity
In all patients the total light dose of 200 J/cm2 on the balloon surface was delivered in 5 fractions alternating laser on/laser off with an off period of 2 min as evaluated in preclinical experiments [16, 28,29,30]. This model allows delivery of photodynamic therapy fluence of 25 J/cm2 at 5 mm inside surrounding brain tissues as described in a previous paper [27].
All 10 patients underwent an intraoperative MRI (iMRI) using a high-field General Electric Medical System Optima 450MRw machine to assess the initial extent of resection (EOR) before the PDT procedure. The iMRI examination included 3D gadolinium-enhanced T1 (T1Gd) and Fast Imaging Employing Steady-state Acquisition (FIESTA) diffusion and perfusion sequences. In cases where residual contrast enhancement was amenable to further resection, a second stage of microsurgical resection was performed based on the iMRI findings. Once this step was completed, the team proceeded to iPDT illumination per trial protocol. This is expected to induce cell death in any remaining tumour cells.
Surgical characteristics are detailed in Table 2. Five patients underwent total resection of contrast enhancement confirmed by the first intraoperative MRI. Only one of them presented with unresectable vague residual fluorescence in the peri cavitary cerebral parenchyma. Four patients underwent a second stage of microsurgical resection after the first intraoperative MRI. Two patients presented with residual vague fluorescence in eloquent areas with further resection deemed unsafe. Only one patient presented with intense residual fluorescence (no supplementary resection due to eloquent localization).
Table 2 Surgical characteristics and iPDT durationAt the end of the resection, the surgical cavity was illuminated with a laser using the dedicated illumination device. The duration of the intraoperative PDT procedure (illumination of the surgical cavity) varied according to the volume of the surgical cavity. It ranged from 15 to 25 min with a median total duration of 18 min. Median device setup time was 14 min, for a median total procedure time of 36 min.
Adjuvant therapy: the Stupp protocolAfter surgery and intraoperative PDT, patients integrated our usual neuro-oncological management: diagnosis confirmation and supplementary information on the standard of care (SOC) for adjuvant treatment. They all accepted and underwent the SOC i.e. concomitant radio-chemotherapy and EANO guidelines [31].
As described in Table 3, all but 1 patient (#09) completed the initial regimen of radiotherapy and concomitant TMZ.
Table 3 Stupp protocol adherenceFive patients completed the full adjuvant TMZ treatment, 4 patients discontinued maintenance TMZ due to disease progression. Patient #09 did not complete maintenance TMZ due to cytopenia.
Patients assessmentAt least 15 visits were scheduled during patient follow-up. Patients were assessed within 6 weeks of intraoperative PDT then monthly, for 9 months, for medical follow-up and adjuvant therapy, then quarterly until tumour recurrence. Each visit included a full clinical examination and a contrast enhanced MRI with gadolinium-enhanced T1 (T1Gd) and T2/FLAIR sequences to assess treatment response according to the RANO criteria [32].
To evaluate HRQOL and brain tumour-related symptoms, the European Organization for Research and Treatment of Cancer QLQ-30 [33] and QLQ-BN20 [34] questionnaires were used. Participants were asked to complete both questionnaires at the start of the study and every three months for 30 months or earlier if they had a recurrence. HRQOL scores were calculated according to the procedures recommended by EORTC.
The QLQ-C30 questionnaire contains 30 items covering global health, physical status, activity limitation, cognitive, emotional, and social functioning, and occurrence of common symptoms related to cancer or treatment. Raw scores are linearly transformed to 0–100 scales. A high score indicates a better global health, functional status, and greater overall quality of life. A 10% change indicates a significant clinical evolution, and a 20% change will be considered major.
The QLQ-BN20 questionnaire is a 20-item self-reporting tool designed as a supplement to QLQ-C30, to assess HRQOL specifically in brain tumour patients. It includes 20 items assessing disease symptoms (headache, seizures, fatigue, hair loss, skin itching, leg weakness, and bladder control problems) and multi-item scales assessing future uncertainty, visual disturbances, motor dysfunctions, and communication deficits. QLQ-BN20 scores are linearly transformed to a 0–100 scale. A high score indicates greater severity of brain tumour-related symptoms.
For the QLQ-C30 and QLQ-BN20 questionnaires, we calculated the change in the number of points in each score between the first and last questionnaires completed by each patient and then averaged the change in each score across all patients for the duration of the study.
For the safety assessment, all adverse events (AEs) and serious adverse events (SAEs) were collected, evaluated, and graded at each visit. Adverse device effects (ADEs) and serious adverse device effects (SADEs) were also collected during surgery and patient hospitalization. AE and ADE intensity was graded according to NCI-CTCAE V5.0 (Common Terminology Criteria for AEs v5.0).
Special attention was paid to AEs related to 5-ALA HCl (such as emesis, nausea, hepatobiliary disorders, haematological disorders, photo sensibilization) and postoperative complications (such as neurological deficit, severe infection, epileptic status, haemorrhage). AEs and SAEs related to the device or to the PDT procedure were recorded.
Statistical analysisFor this pilot feasibility study, the number of subjects was not based on statistical assumptions. A total of 10 evaluable patients were enrolled in the study, with the aim that at least 60% of the enrolled patients (i.e., 6 out of 10) would be able to undergo the complete intraoperative PDT without unacceptable and/or unexpected toxicities.
The study is now completed, and the database was locked in September 2021. Five years were calculated from the last included patient and November 2023 was the cut-off date for collecting survival data for the patients still alive.
We have compiled the safety results described in the previous publication [26] and analysed the efficacy and quality of life results until relapse or November 1st, 2023, whichever came first.
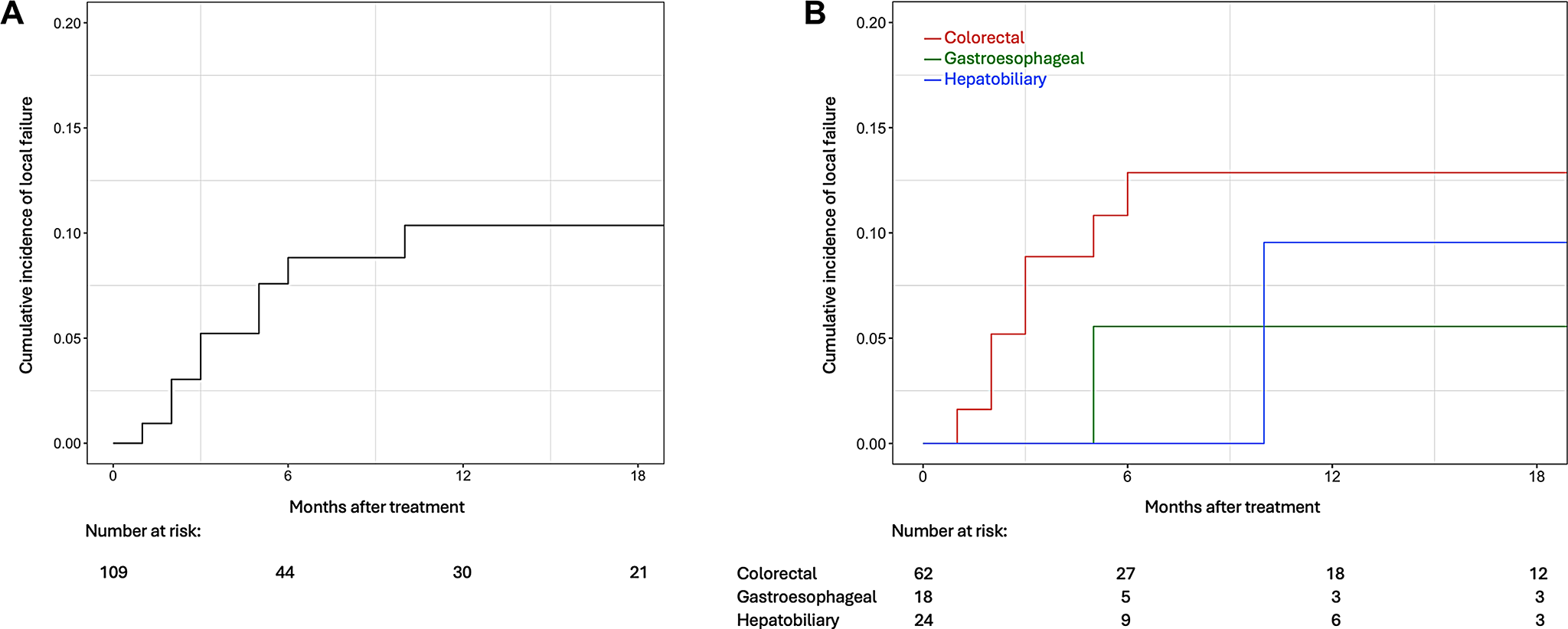
Survival analysis from diagnosis to tumour progression, death or last follow-up was plotted using the Kaplan–Meier method (performed using a graphic user interface to the R statistical analysis software for scientific publications, Medistica©, pvalue.io, 2021). Patients were censored at the time of last follow-up or at the time of death.
Comments (0)