
Remember me

C81 was kindly provided by the group of Prof. Franz Bracher (Pharmaceutical and Medicinal Chemistry, Department of Pharmacy – Center for Drug Research, Ludwig-Maximilians-Universität München, Munich, Germany) with a purity of 98.4% [12, 13]. The CLK inhibitors MU1210 and T3-CLK were purchased from Sigma-Aldrich (St. Louis, Missouri, USA) or were kindly provided by the group of Prof. Stefan Knapp (SGC, Institute of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt, Germany). The DYRK2 inhibitor LDN-192,960 and the glycogen synthase kinase-3 (GSK3) inhibitor LY2090314 were purchased from MedChemExpress (Monmouth Junction, New Jersey, USA); the PIM kinase inhibitor AZD1208 was obtained from Selleckchem (Houston, Texas, USA). The chemical structures of C81, MU1210, and T3-CLK can be found in Supp. Figure 1. All kinase inhibitors, including C81, were dissolved and aliquoted in DMSO (Sigma-Aldrich) at concentrations ranging from 10 to 100 mM, adjusted for solubility to achieve the lowest possible DMSO concentrations in the final cell culture experiments and stored at − 80 °C until use. In cell culture and ex vivo experiments, DMSO concentrations never exceeded 0.1%, whereas in in vivo animal experiments, DMSO concentrations never exceeded 0.01% in the eye.
AnimalsAll in vivo animal procedures were conducted in compliance with protocols approved by the local governmental authorities (Tierschutzkommission acc. § 15 TSchG of the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, with the permission number Az 81-02.04.2022.A338) and were in accordance with the National Institutes of Health (NIH Bethesda, Maryland, USA) guidelines. Mice were housed in individually ventilated caging (IVC) systems (GM 500, Tecniplast Greenline) with a maximum cage density of five adult mice per cage. Light was adjusted to a 12-h/12-h light/dark cycle with light on at 6 am, temperature and relative humidity were regulated to 22 ± 2 °C, and 45–65% relative humidity. Mice were fed irradiated phytoestrogen-free standard diet for rodents (Altromin 1314; 59% carbohydrates, 27% protein, 14% fat) and had access to food and acidified water ad libitum. 8- to 10-week-old C57BL/6J mice with an averaged body weight of 19 g ± 1.5 g for females and 25 g ± 2 g for males were used for experiments.
Ex vivo animal procedures were performed in compliance with the German Animal Welfare Act (§ 4 Tierschutzgesetz) and approval number V54-19c20/21I-FR/Biologicum, Tierhaus Campus Riedberg (Regierungspräsidium Darmstadt, Germany). The ex vivo animal experiments were conducted using 4- to 6-week-old C57BL/6 N mice, which were kept under a 12-h/12-h light/dark cycle and had access to food and water ad libitum until sacrifice.
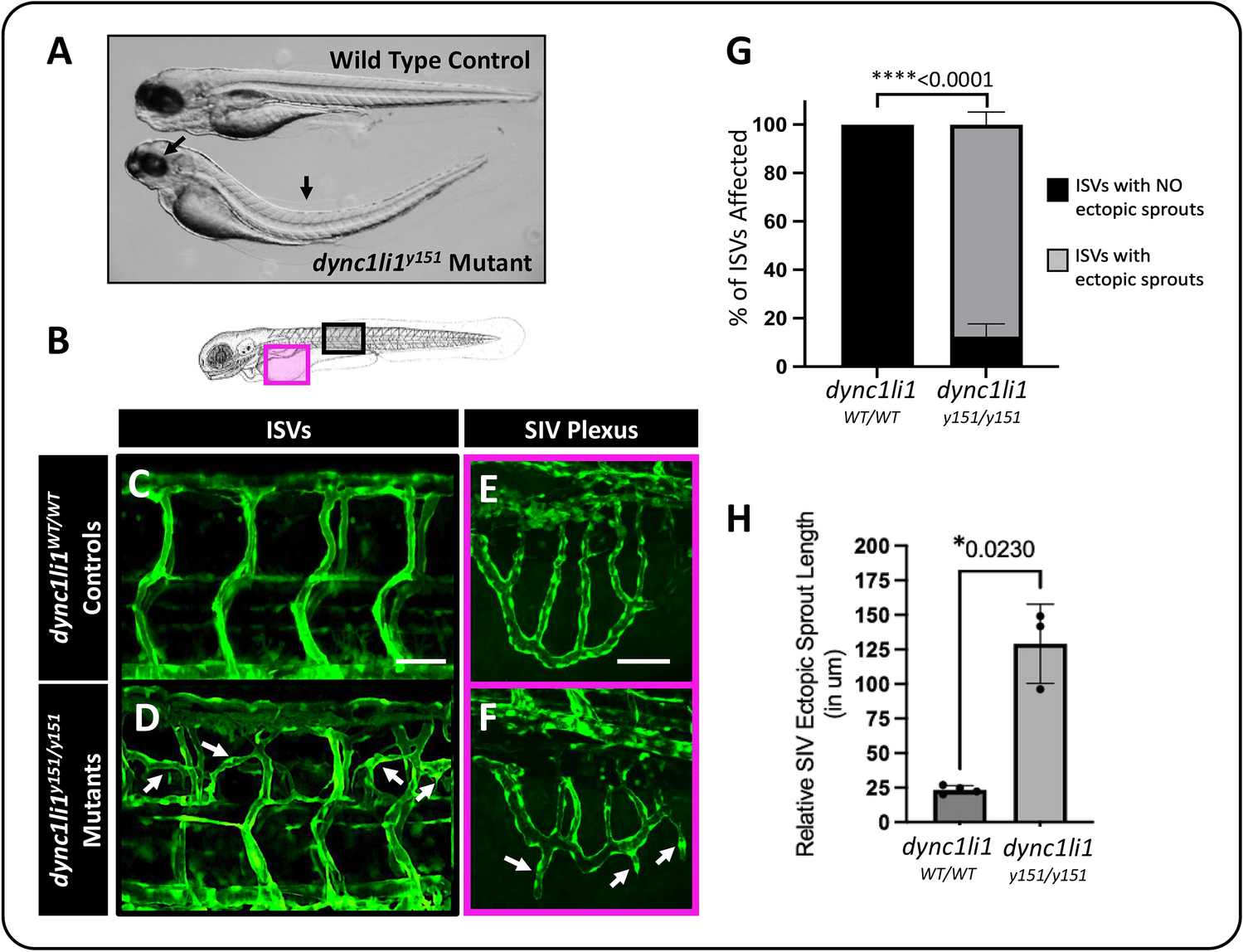
Laser photocoagulationLaser photocoagulation was carried out as described previously [22]. In brief, mice were anesthetized with a mixture of ketamine (100 mg/kg body weight, Ketavet) and xylazine (5 mg/kg body weight, 2% Rompun) diluted in 0.9% sodium chloride by intraperitoneal (i.p.) injection. Their pupils were dilated with a topical drop of phenylephrine 2.5%/tropicamide 0.5%. A slit lamp-mounted diode laser system (Quantel Medical Vitra, 532-nm green laser, power 100 mW, duration 100 ms, and spot size 100 μm) was used to generate three equal laser burns around the optic nerve in each eye with a cover glass as a contact lens. To validate rupture of Bruch’s membrane, infrared (IR) images were recorded using Spectralis HRA/OCT device to analyze post-laser retinal structure and laser lesion size in vivo. Exclusion criteria were cataract and corneal epithelial edema before laser photocoagulation, unsuccessful laser burns without Bruch’s membrane rupture, or severe choroidal hemorrhages.
Intravitreal drug administrationThe animals were randomly assigned to the experimental groups. The following compounds (all diluted in phosphate-buffered saline, PBS) were injected intravitreally immediately after laser pulse application. 1 µl of either 15-µM or 50-µM C81 or corresponding vehicle controls (0.015% or 0.05% DMSO) were applied to reach 3 µM or 10 µM final concentration assuming 4-µl vitreous volume [23]. Therefore, eyes were treated with oxybuprocaine (Conjuncain, 0.4 mg/ml) eye drops and a 34-gauge needle was inserted into the vitreous space approximately 1.5 mm below the limbus and the compounds were administered bilaterally with a NanoFil syringe (Word Precision Instruments, Sarasota, FL, USA). Afterward, eyes were covered with Bepanthen eye and nose ointment (Bayer, Leverkusen, Germany).
Fundus photography and fundus fluorescein angiography (FFA)Vascular leakage was analyzed 3 and 7 days after laser photocoagulation. After anesthesia and pupil dilatation, mice received i.p. injection of 0.1 ml of 2.5% fluorescein diluted in 0.9% sodium chloride. Late-phase angiograms were recorded 10 min after fluorescein injection using Spectralis HRA/OCT. Simultaneously, IR fundus images were acquired to analyze the laser lesion size. The size of laser lesions and vascular leakage was determined using the measuring tool of the HEYEX software (Heidelberg Engineering, Heidelberg, Germany). The analysis of vascular leakage by measuring pixel intensities was performed as described previously [24].
Immunohistochemistry of retinal and RPE/choroidal flat mountsMice were euthanized by cervical dislocation and the eyes enucleated and fixed in 4% of Roti-Histofix (Carl Roth, Karlsruhe, Germany) for 3 h at room temperature (RT). The dissected RPE/choroidal flat mounts were permeabilized and blocked overnight in Perm/Block buffer (5% normal donkey serum (NDS), 0.2% bovine serum albumin (BSA), 0.3% Triton X-100 in PBS) at 4 °C. RPE/choroidal flat mounts were stained in addition with FITC-conjugated isolectin B4 from Bandeiraea simplicifolia (BS, 1:100 diluted in Perm/Block, L2895, Sigma-Aldrich). After several washing steps, retinal and RPE/choroidal flat mounts were mounted on a microscope slide and embedded with fluorescence mounting medium (Vectashield HardSet H-1400, Vector Labs, Newark, California, USA). Images were taken with a Zeiss Imager. M2 equipped with an ApoTome.2 (Carl Zeiss, Oberkochen, Germany). Areas of CNV in RPE/choroidal flat mounts were measured with the spline function of the graphic tool included in the ZEN software (Zeiss). The average CNV area per eye was calculated.
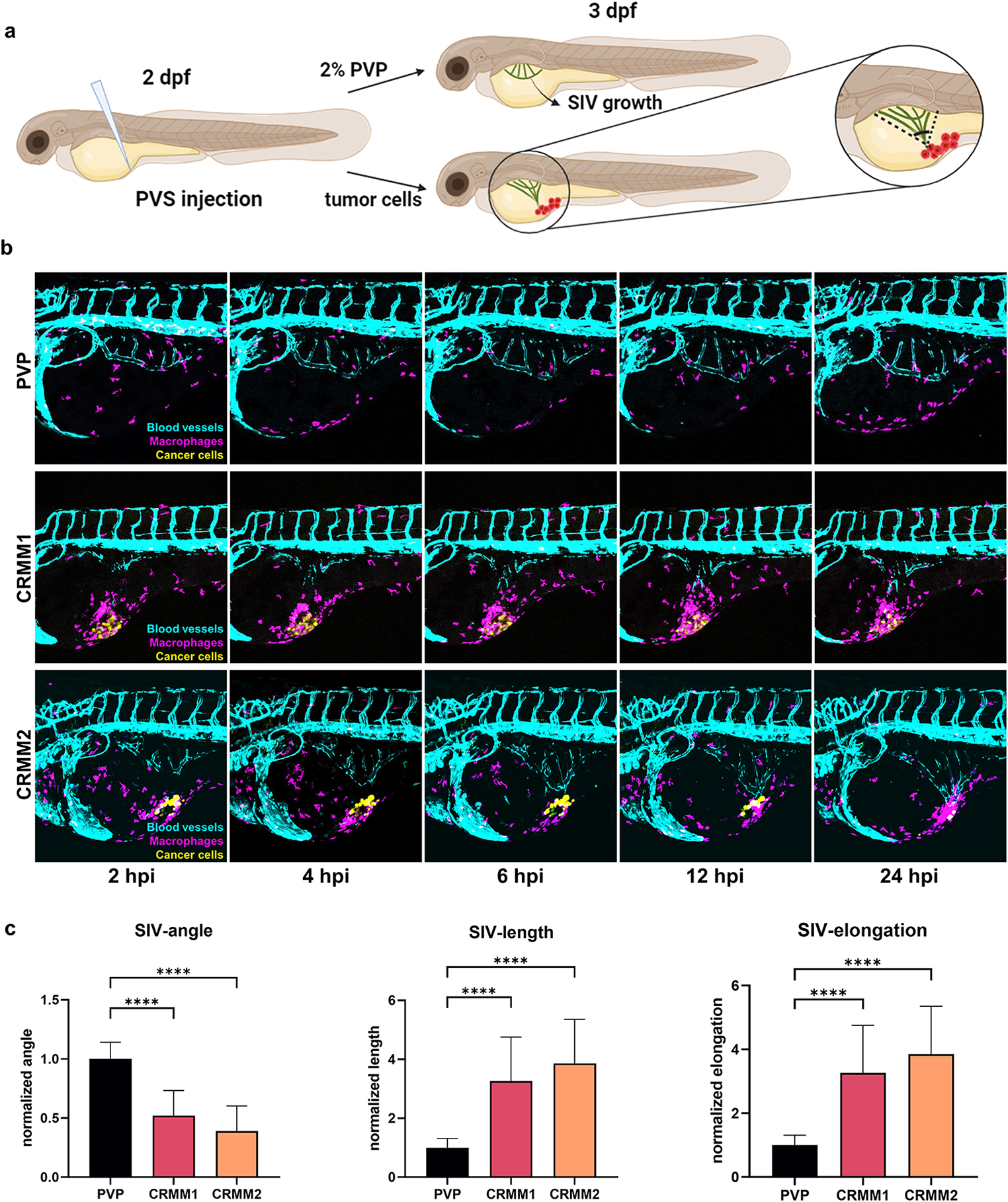
Ex vivo mouse aortic ring assayMouse aortic ring assays were performed with 4- to 6-week-old C57BL6/N mice, which were kindly provided by the group of Prof. Achim Schmidtko (Institute of Pharmacology and Clinical Pharmacy, Goethe University Frankfurt, Frankfurt, Germany), as previously described [25, 26]. Briefly, mice were sacrificed using CO2. Death was ensured by subsequent breaking of the necks. Aortae were explanted, cleaned of surrounding tissue, and cut into rings of about 0.5–1 mm length. These rings were then incubated overnight in Opti-MEM I (Gibco/Thermo Scientific, Waltham, Massachusetts, USA) supplemented with 100-U/ml penicillin and 100-µg/ml streptomycin (P/S; Pan-Biotech, Aidenbach, Germany). On the following day, rings were embedded into a 50-µl rat tail collagen I gel (1.5 mg/ml in M199; Corning, Corning, New York, USA) and incubated with 150 µl of Opti-MEM I supplemented with P/S, 2.5% FCS Superior (Sigma-Aldrich), and 30-µg/ml murine vascular endothelial growth factor (mVEGF165; PeproTech, Rocky Hill, New Jersey, USA) until first endothelial sprouts were visible (3–5 days). Afterward, sprouting rings were treated with 3-µM C81 or vehicle control for 3 additional days. Subsequently, the stimulation was terminated by fixating the rings with ROTI-Histofix (Carl Roth) for 30 min. To stain the rings, they were first permeabilized using 0.25% Triton X-100 (Carl Roth) twice at room temperature. Unspecific binding sites were blocked using 1% BSA (Carl Roth) in PBS at 4 °C overnight, followed by staining with FITC-coupled BS-I Lectin (L9381, 0.1 mg/ml, Sigma-Aldrich) and a CY3-conjugated antibody against smooth muscle actin (α-SMA, C6198, Dilution 1:1000, Sigma-Aldrich) at 4 °C overnight. After thorough washing of the rings with 0.1% Triton X-100, images were taken using a confocal laser scanning microscope (LSM 780, Zeiss), and sprouting was quantified manually using Fiji/ImageJ (version 1.53t, NIH).
Cell cultureHUVECs were bought from PELOBiotech (Martinsried, Germany) or were isolated from human umbilical cords of anonymized, healthy donors as previously described by Jaffe et al. [27] (the research Ethics Committee/Institutional Review Board approved the waiver W1/21Fü for the use of anonymized human material on September 15th, 2021). Briefly, the umbilical veins were washed with warm PBS including Ca2+ and Mg2+ (PBS+) to remove remaining cord blood and then incubated with a collagenase A solution (0.1 g/l; Roche, Basel, Switzerland) for 45 min at 37 °C. Afterward, cells were detached from the vessel walls by gentle tapping on the outside of the umbilical cords, flushed out using warm M199 (PAN-Biotech) supplemented with 10% FCS (Sigma-Aldrich) and P/S (PAN-Biotech) and collected. Subsequently, the cells were pelleted by centrifugation at 300 g for 5 min, the supernatant was discarded, and cells were resuspended in endothelial cell basal medium (PELOBiotech) containing 10% FCS (Sigma-Aldrich), P/S (PAN-Biotech), 2.5-µg/ml amphotericin B (PAN-Biotech), and EASY endothelial cell growth supplement (PELOBiotech). This medium will henceforth be called fully supplemented ECGM. Cell suspensions were then seeded on 25 cm2 cell culture flasks (Sarstedt, Nümbrecht, Germany) coated with collagen G (Sigma-Aldrich). HUVECs were generally split every 2–4 days at a ratio of 1:3, expanded until passage 2 and, for experimental purposes, exclusively used in passage 3. HMEC-1, a microvascular cell line [28], were obtained from the CDC (lot 119223; Centers for Disease Control and Prevention, Atlanta, Georgia, USA) and cultivated on collagen G-coated 25 cm2 or 75 cm2 flasks (Sarstedt) using fully supplemented ECGM (PELOBiotech). They were split every 2–3 days at a ratio of 1:3 and used up to passage 30. Stably transfected HMEC-1 were only used until passage 15.
Spheroid sprouting assayHUVECs or HMEC-1 spheroids were generated using the hanging drop method, as previously described [29]. Briefly, 400 HUVECs or HMEC-1 were seeded as droplets onto square petri dishes (Greiner Bio-One, Kremsmünster, Austria), which were flipped upside down and incubated for 24 h. Spheroids were then collected from the droplets by flushing with PBS+, washed, and then embedded in a rat tail collagen I gel (1.5 mg/ml in M199, Corning) containing methylcellulose (Sigma-Aldrich) and 5% FCS (Sigma-Aldrich). After collagen polymerization, the spheroids were treated with the indicated compounds at the indicated concentrations for 30 min. Subsequently, sprouting was induced using human recombinant VEGF165 (PeproTech) at 10 ng/ml for 20 h, after which spheroids were fixed using ROTI-Histofix (Carl Roth) for 30 min. Finally, images were taken using a Leica DMI IL LED inverted microscope (Leica Microsystems, Wetzlar, Germany) and analyzed manually using Fiji/ImageJ (NIH).
Proliferation assay1,500 cells (HUVECs or HMEC-1) per well were seeded on collagen G-coated 96-well plates (Greiner Bio-One) and grown in fully supplemented ECGM (PELOBiotech) for 24 h. Afterward, cells were either washed, fixated using methanol/ethanol (2:1) for 10 min, and then stained using crystal violet (Carl Roth), or treated with the indicated concentrations of C81 or vehicle control for another 72 h. The incubation of the treated cells was then stopped as described above. After staining with crystal violet, cells were washed using water until the water ran clear. Crystal violet was leached using 20% acetic acid, and absorbance was measured using a plate reader (Tecan, Männedorf, Switzerland). Cells fixated after 24 h were used for baseline normalization.
Scratch assayUndirected migration was studied using a scratch assay. For this, cells (HUVECs or HMEC-1) were seeded on 24-well plates (Greiner Bio-One) and grown to confluency. Consequently, a scratch was introduced to the monolayer using a 10-µl XL pipette tip (Greiner Bio-One), after which cells were washed and treated with the indicated concentrations of the indicated compounds, a vehicle control, and a serum starvation control (1% FCS in M199). Cells were allowed to migrate for 9 to 12 h or until the scratches in the vehicle control were mostly closed. Subsequently, images were taken using a DMI IL LED inverted microscope (Leica Microsystems) and quantified using Fiji/ImageJ (NIH). Serum starvation controls served as a baseline.
Boyden chamber assay100,000 cells (HUVECs) were seeded on collagen G-coated cell culture inserts (Corning) made from polycarbonate with a pore size of 8 μm and left to adhere for 2–3 h. Subsequently, cells were treated with the indicated concentrations of C81, introduced to a 0–20% FCS gradient (0% FCS in the upper chamber, 20% in the lower chamber) in M199, and allowed to migrate for 16 h. Afterward, the cells were washed with PBS, fixated using methanol/ethanol (2:1), stained with crystal violet, and then washed again until PBS ran clear. Cells that did not migrate were removed from the top of the insert by gently scraping them off with a cotton swab. Finally, remaining crystal violet was leached using 20% acetic acid, and absorbance was measured using a plate reader (Tecan).
Live-cell chemotaxis assay2D chemotaxis of scarcely seeded HUVECs was evaluated using µ-Slide Chemotaxis slides (ibidi, Martinsried, Germany) according to the manufacturer’s instructions. For this, 18,000 HUVECs were seeded onto the channel of the slide and left to adhere for 2 h. Afterward, cells were washed with M199 (Sigma-Aldrich) containing P/S (PAN-Biotech), treated with 10-µM C81 or vehicle control and subjected to an FCS (Sigma-Aldrich) gradient (0–20%). Subsequently, cells were allowed to migrate for 20 h in a climate chamber (5% CO2, 37 °C), and microscopic images were taken every 10 min on a DMI6000 B microscope (Leica Microsystems). Migration was analyzed using the Manual Tracking and Chemotaxis Analysis plugins for Fiji/ImageJ (NIH).
Tube formation assayTube formation was measured using a Matrigel (Corning) based assay. Initially, wells in a µ-slides Angiogenesis (ibidi) were coated with 10-µl growth factor-reduced Matrigel (Corning). Subsequently, 10,000 HUVECs per well were seeded onto the Matrigel in fully supplemented ECGM containing the indicated treatments. HUVECs were allowed to form tubes for 7.5 h, after which images taken immediately using a Leica DMI6000 B inverted microscope. Images were then quantified for number of junctions and number of master segments using the angiogenesis analyzer plugin for Fiji/ImageJ (NIH).
SDS-PAGE and western blot analysisHUVECs or HMEC-1 were grown to confluency and treated as indicated. Unless specified otherwise, treatments were performed in fully supplemented ECGM. After the incubation times were over, cells were washed with cold PBS and lysed using a RIPA buffer containing PMSF (Roche), NaF (Sigma-Aldrich), EDTA-free Complete Mini (Roche), and Na3VO4 (Sigma-Aldrich). If phosphoproteins were analyzed, the lysis buffer additionally contained phosphatase inhibitors. Afterward, the protein concentrations of the lysates were determined using a bicinchoninic acid assay kit (Thermo Scientific) according to the manufacturer’s instructions. Subsequently, a Tris–HCl (Carl Roth)-based buffer containing pyronin Y (Sigma-Aldrich), sodium dodecyl sulfate (SDS, Sigma-Aldrich), glycerol (Carl Roth), and dithiothreitol (DTT, Sigma-Aldrich) were added to each sample, and proteins were denatured by heating the samples to 95 °C for 5 min. Consequently, 20–25 µg of protein was run on a polyacrylamide gel (between 7.5 and 15%, depending on the analyzed proteins, Carl Roth) and blotted onto a 0.2-μm polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, California, USA) using a Transblot Turbo device (Bio-Rad) or by tank blotting at 30 V for 16 h or 100 V for 1 h. Unspecific binding sites were then blocked using BSA (Carl Roth) or non-fat dried milk (Blotto, Carl Roth), both at 5% in TBS containing 0.1% Tween 20. Finally, the membranes were incubated with antibodies for 2 h at room temperature (RT) or overnight at 4 °C, both conditions with gentle shaking, and visualized using luminol-based enhanced chemiluminescence (ECL) and X-ray films (Fujifilm, Tokyo, Japan) or a ChemiDoc XRS+ (Bio-Rad) imager. The images were quantified using the densitometry feature of Fiji/ImageJ (NIH). When two or more proteins of similar sizes were analyzed on the same membrane, previous antibodies were stripped off the membrane by incubating the membrane for 20 min using an acidic (pH 2.2) stripping buffer containing glycine (Carl Roth), 0.1% SDS (Sigma-Aldrich), and 1% Tween 20 (Carl Roth). Afterward, membranes were washed, and unspecific binding sites were blocked again before membranes were incubated with the next antibody. The following primary antibodies were used: anti-VEGF receptor 2 (55B11) rabbit mAb #2479 (dilution 1:2000; Cell Signaling Technology, CST, Danvers, Massachusetts, USA); anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) mouse mAb #9106 (dilution 1:2000; CST); anti-p44/42 MAPK (Erk1/2) antibody #9102 (dilution 1:1000; CST); anti-phospho-AKT (Ser473) rabbit mAb #4060 (dilution 1:2000, CST); anti-AKT (pan) rabbit mAb #4691 (dilution 1:2000, CST); mouse monoclonal anti-β-actin peroxidase-linked antibody A3854 (dilution 1:50.000, Sigma-Aldrich); mouse anti-phospho-epitope SR proteins antibody, clone 1H4 MABE50 (dilution 1:1000; Sigma-Aldrich); and anti-β-catenin (D10A8) XP rabbit mAb #8480 (dilution 1:2000; CST). Secondary antibodies were goat anti-rabbit IgG, HRP-linked antibody #7074 (dilution 1:3000; CST) and horse anti-mouse IgG, HRP-linked antibody #7076 (dilution 1:3000; CST).
Quantitative real-time PCR (qPCR)To assess relative mRNA expression, quantitative real-time PCR was performed. For this, cells were initially subjected to the indicated conditions, after which total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, including on-column DNAse digestion. 1 µg of isolated RNA was then reverse transcribed into cDNA using FastGene Scriptase II (Nippon Genetics Europe, Düren, Germany) with random hexamer primers (New England Biolabs NEB, Ipswich, Massachusetts, USA) according to the manufacturer’s protocol. cDNA was then diluted 1:25, and gene expression was quantified using the primers stated in Table 1 and the SyGreen Blue Hi-ROX Mastermix (PCR Biosystems, London, United Kingdom) on a StepOne Plus device (Applied Biosystems/Thermo Scientific). Relative Gene expression was quantified with the ΔΔCt method using GAPDH as the control gene.
Table 1 Primers used for qPCRKinase assayAffinity for C81 against CLKs was measured by Eurofins DiscoveRx (San Diego, California, USA) using their KdELECT Assay Platform in 11 concentrations ranging from 0.1 to 10 µM in duplicates.
Assessment of interchromatin granule clusters (IGCs)55,000 HUVECs were grown on collagen G-coated 8-well slides with coverslip bottoms (ibidi) until confluency and then incubated with the indicated concentrations of the compounds for 6 h. Afterward, cells were washed with cold PBS and fixated using ROTI-Histofix (Carl Roth) for 10 min. Next, the fixated HUVECs were permeabilized using 0.1% Triton X-100 (Carl Roth), and unspecific binding sites were blocked using 1% BSA (Carl Roth) in PBS. Subsequently, the same antibody against phosphorylated SR proteins as used for western blotting (MABE50, dilution 1:500; Sigma-Aldrich) was applied to stain IGCs and visualized using a secondary anti-mouse antibody coupled to Alexa 488 (dilution 1:400; Thermo Scientific). Hoechst 33342 (Sigma-Aldrich) served as a control stain of the nuclei. Images were taken using a Leica DMI6000 B epifluorescence microscope (Leica Microsystems).
siRNA transfection and knockdownsHUVECs were transfected with siRNAs targeting CLK1, CLK2, CLK3, CLK4, β-catenin (CTNNB1) or a non-targeting control (all Dharmacon ON-TARGET Plus SMARTpools, Horizon Discovery, Lafayette, Colorado, USA) using either Lipofectamine RNAiMAX (Thermo Scientific) or GeneTrans II (MoBiTec, Goettingen, Germany) according to the manufacturer’s instructions. When Lipofectamine RNAiMAX was used, HUVECs were transfected with 25-pmol siRNA per well in a 6-well plate for 24 h and afterward incubated or treated as indicated. When Genetrans II was used, HUVECs were transfected with 80-pmol siRNA per well in a 6-well plate, prediluted in Diluent B, for 4 h, and afterward treated as indicated. Unless otherwise specified, incubation timepoints are counted from the beginning of the transfection. Analysis was performed using the previously described spheroid sprouting assay, SDS-PAGE with subsequent western blot analysis, or qPCR.
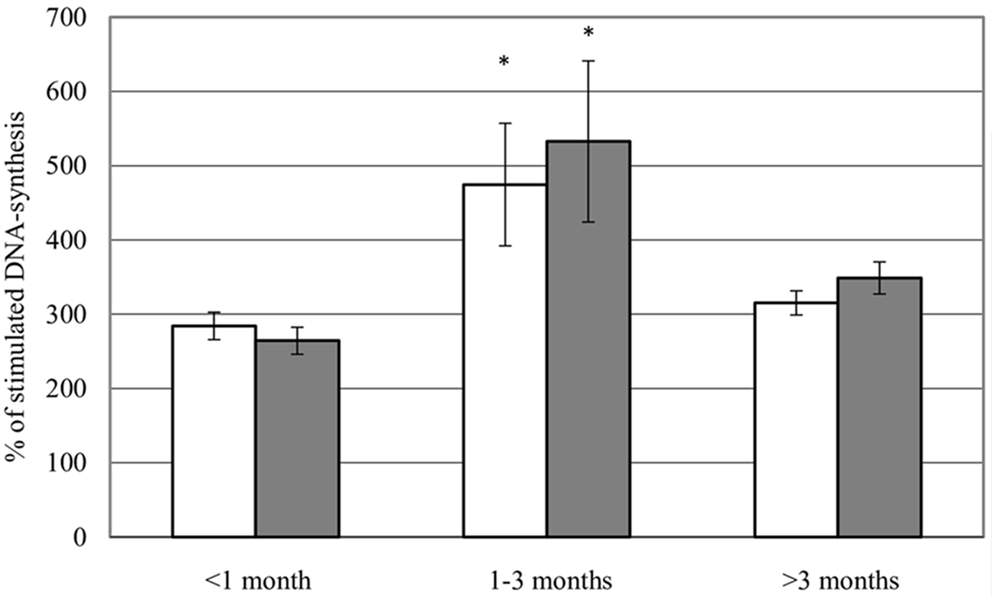
Propidium iodide staining and flow cytometryApoptosis was measured using the method described by Nicoletti et al. [30]. Briefly, HUVECs were treated with the indicated concentrations of MU1210, staurosporine (positive control), or vehicle control for 24 h. Afterward, cells were washed and detached from the culture vessels; supernatants and all washing solutions were conserved. Subsequently, the cells were centrifuged at 300×g and 4 °C for 10 min and stained using a hypotonic fluorochrome solution (HFS) containing Triton X-100 (Carl Roth) and propidium iodide (Carl Roth) for 24 h at 2–8 °C. Consequently, the fluorescence intensity of single cells was analyzed using a flow cytometer (FACSVerse, BD, Franklin Lakes, New Jersey, USA). Doublets were removed through gating; lower fluorescence intensity than typical for G0/G1 phase cells indicated DNA degradation associated with apoptosis. Alternatively, propidium iodide staining was used to determine cell cycle distribution of proliferating HUVECs. When this was done, 44,000 HUVECs were seeded in a well of a 6-well plate in fully supplemented ECGM and left untreated for 24 h, after which they were treated as indicated. 48 h later, cells were washed, detached, stained, and measured as described above. Cell cycle distribution was determined from the fluorescence intensity of the cells.
RNA sequencing (RNA-Seq)To generate hypotheses about potentially affected pathways as well as alternative splicing events, RNA-Seq was performed. For this, confluent HUVECs were treated with 10-µM C81, MU1210, or a vehicle control for 6 h, after which total RNA was isolated using the RNeasy Micro Kit (Qiagen) according to the manufacturer’s instructions. The quality of the RNA was then assessed using a Tapestation 4150 (Agilent, Santa Clara, California, USA), and libraries were prepared using the Lexogen Corall Total RNA-Seq Kit (Lexogen, Vienna, Austria) with the poly-A selection module and the qPCR module according to the manufacturer’s instructions. The quality of the libraries was verified using a Tapestation 4150 (Agilent) and quantified using a Qubit 3 Fluorometer (Thermo Scientific). Libraries were then sequenced on a NextSeq 2000 Device (Illumina, San Diego, California, USA) using paired end reads of 105 bp length and approximately 50 million reads per sample. Post-processing of reads (quality control, trimming, alignment) was done using the BlueBee platform included with the library prep kit.
Gene ontology (GO) term analysisDifferentially expressed genes were detected from the RNA-Seq data using DESeq2 (Version 1.36.0) for R (version 4.2.1) [31]. Subsequently, downregulated genes, as defined by an adjusted p value of 0.05 or smaller and a log2 fold change ≤ − 0.5, were subjected to a GO term analysis using clusterProfiler (version 4.4.4) for R [32]. Adjusted p values of 0.05 or smaller were considered statistically significant; graphs were created using ggplot2 for R [33].
Alternative splicingAlternatively spliced exons were detected from RNA-Seq data using rMATS Turbo 4.1.2 for python 3.9 with the settings: read length 105, paired end reads, allow detection of novel splice sites, forbid clipping, and allow variable read lengths [34]. Results were filtered for relevant splicing events by coverage (at least 20 total junction reads per event), statistics (false discovery rate of 0.05 or smaller was considered statistically significant), and by the difference in the inclusion level (a difference of 0.1, which equates to 10%, was considered relevant) using dplyr (version 1.1.2) for R, and results were visualized using dot plots from ggplot2 (R) or Sashimi plots created with rmats2sashimiplot (version 2.0.4; python 2.7) [33, 35]. Afterward, alternatively spliced genes were subjected to a GO term analysis as described above. The lists of alternatively spliced genes were also scanned for WNT-associated genes using the PANTHER database (Version 17.0) through the web interface [36, 37].
Endpoint PCRAlternative splicing of CLK1 was additionally investigated using endpoint PCR to verify sequencing results. For this, HUVECs were treated with 10-µM C81, MU1210, or vehicle control for 6 h, after which RNA was isolated and reverse transcribed to cDNA as described above. Subsequently, cDNA was subjected to endpoint PCR using GoTaq DNA Polymerase (Promega, Wisconsin, USA) according to the manufacturer’s instructions with 35 cycles of the following program: initial denature: 95 °C, 2 min; denature 95 °C 1 min; annealing 56 °C 1 min; extension 72 °C 30 s; and final extension 72 °C 5 min. The length of the amplicons was then determined using 3% agarose gels (Thermo Scientific) supplemented with ethidium bromide (Carl Roth) and using a 100-bp ladder (NEB) as a reference. After electrophoresis in TBE buffer, DNA was visualized in the gel with an Azure C200 (Azure Biosystems, Dublin, California, USA) gel imaging system. Primers can be found in supplementary Table 1; an amplicon length of 177 bp indicated exon skipping and a length of 268 bp indicated exon inclusion.
CloningThe TCF/LEF and delTCF/LEF reporter gene plasmids were cloned into a sleeping beauty transposon backbone [38], kindly provided by Prof. Rolf Marschalek (Institute of Pharmaceutical Biology, Goethe University Frankfurt, Frankfurt, Germany), using the promoter, TCF/LEF response elements and luciferase sequence of the pGL4.49 Plasmid (Promega). For delTCF/LEF plasmids, the response elements were deleted from the sequence. The backbone contains an eGFP sequence behind a constitutive promoter, which serves as a transfection and selection control. The promoter region and luciferase sequence of pGL4.49, in addition to the sleeping beauty backbone, were linearized using PCR with the primers found in supplementary Table 1, with an overlap between backbone and inserts. This was done using the Q5 High-Fidelity DNA Polymerase (NEB) using the following program: initial denature: 98 °C, 30 s; denature 98 °C 10 s; annealing 64 °C 30 s; extension 72 °C 1 min for inserts, 3 min 40 s for the backbone; and final extension 72 °C 2 min. The linearized fragments were purified by agarose gel electrophoresis followed by gel extraction (Zymoclean Gel Extraction Kit, Zymo Research, Freiburg, Germany) and subsequently assembled using the NEBuilder Kit (NEB) according to the manufacturer’s instructions. The assembled plasmids were transformed into chemically competent DH10β E. coli cells and cultivated at 37 °C on agarose overnight. On the following day, clones were picked and cultivated overnight in a 2-ml liquid culture at 37 °C, 180 rpm. Plasmids were isolated using the GeneJET Miniprep Kit (Thermo) and were sequenced using Sanger sequencing at Microsynth Seqlab (Tübingen, Germany). Correct clones were grown in 100-ml cultures, and plasmids were isolated using the PureYield Plasmid Midiprep System (Promega). Subsequently, remaining endotoxins were removed using the MiraCLEAN Endotoxin Removal Kit (Mirus Bio, Madison, Wisconsin, USA) according to the manufacturer’s instructions.
Luciferase reporter gene assayHMEC-1 (1 million cells) were transfected with either TCF/LEF or delTCF/LEF plasmids, together with a SB100x transposase plasmid kindly provided by Dr. Eric Kowarz (Institute of Pharmaceutical Biology, Goethe University Frankfurt, Germany), at a ratio of 19:1, using the Nucleofector IIb device (Lonza, Basel, Switzerland) and the AMAXA HUVEC Nucleofector Kit and program A-034 according to the manufacturer’s instructions. For each transfection, 5 µg of total DNA were used. Transfected cells were transferred to a collagen G (Sigma-Aldrich)-coated well of a 6-well plate, cultivated for 2 days in fully supplemented ECGM (PELOBiotech), and subsequently selected using puromycin at 1–3 µg/ml for up to 2 weeks. Transfection and selection efficiencies were routinely checked using a Leica DMI6000 B Fluorescence Microscope (Leica Microsystems, Wetzlar, Germany). After successful selection, cells were seeded onto 48-well plates (Greiner Bio-One) and grown to confluency. Confluent cells were serum starved overnight (1% FCS in M199), then pretreated with the indicated concentrations of C81 for 30 min, and after which β-catenin signaling was induced using the GSK3 inhibitor LY2090314 at 30 nM. After 6 h, the incubation was stopped, and cells were lysed using 65-µl passive lysis buffer (Promega). 10 µl of the lysis solution was transferred to a white 96-well plate (Thermo Scientific). Luminescence was induced using the firefly substrate of the Luciferase Assay System (Promega) and measured using a plate reader (Tecan).
StatisticsGraphs and statistics of all experiments, except RNA-Seq experiments, were created using GraphPad Prism 10.0.2 (Dotmatics, Boston, Massachusetts, USA). Generally, experiments were carried out in 3 or more independent replicates, and statistical significance was calculated using an unpaired 2-tailed students t test (experiments containing 2 groups) or an unpaired 1-way or a 2-way ANOVA (experiments with 3 or more groups). For ANOVA, post hoc analysis was performed to detect significant differences between specific datasets. Unless specified otherwise, this was done using Dunnett’s post hoc test, comparing treatments and negative controls to the single positive control. To improve readability, statistically significant differences, as defined by a p value of 0.05 or smaller, were marked with a single asterisk or other specified symbol, regardless of significance level, whereas non-significant differences are not marked at all.
Comments (0)