Phosphatidylserine (PS) primarily locates in the inner leaflet of cell membrane and this asymmetry is actively maintained by flippases such as ATPase Phosphlipid Transporting 11 A and 11C. Meanwhile, scramblases drive ATP-independent, bidirectional movement of membrane lipids between the extracellular and the cytoplasmic leaflet [[1], [2], [3]]. Scramblase Xk-related protein 8 (Xkr8) is activated by caspases and upregulated [4] during cell apoptosis, leading to irreversible PS externalization on the cell surface which serves as an “eat me” signal for phagocytosis, a process known as efferocytosis [[5], [6], [7]]. Professional phagocytes such as macrophages and dendritic cells express multiple PS-recognizing receptors, including T cell immunoglobulin mucin domain (TIM) family (TIM-1, −3, and − 4) [[8], [9], [10]], tyrosine kinase family (Axl, Tyro3, and MerTK) [11,12], scavenger receptor family (SCARF1) [13], and integrins, which can trigger profound immunosuppressive effects upon interaction with PS-exposing cells [[14], [15], [16]]. After engulfing apoptotic cells, macrophages can be induced to polarize toward M2-like phenotype, increasing the secretion of anti-inflammatory cytokines interleukin-10 (IL-10) and TGF-β, which is crucial to maintain homeostasis in normal tissue [17,18].
In the context of tumors, the interaction between PS+ apoptotic cells and macrophages promotes the accumulation of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) while inhibiting the priming of tumor-specific adaptive immune response in the tumor microenvironment (TME) [19]. Immunosuppressed TME poses significant challenges in cancer therapy, facilitating immune evasion, drug resistance and tumor progression [20]. Various efforts have been made to modulate TME in the development of immunotherapies, including checkpoint inhibitors targeting PD-1/PD-L1 and CTLA-4 [[21], [22], [23], [24]]. Recently, PS has emerged as a new checkpoint and critical target in counteracting immunosuppressive TME [17,25]. Strategies targeting PS and related signaling pathways such as Annexin A5, and PS- and MerTk-specific antibodies, have been developed to counteract its immunosuppressive effects [26,27]. However, despite the initial encouraging results in improving anti-tumor immunity and therapeutic efficacy in preclinical and early phase clinical studies [28], PS antibodies failed to show clear benefit over chemotherapy [14]. In addition, there are safety concerns that targeting PS across the whole body may impair its normal physiological function in non-cancerous tissues, suggesting another hurdle for translational application [27].
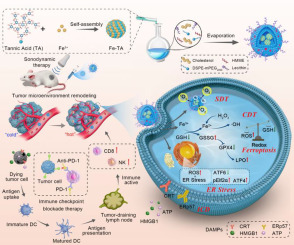
We have recently proposed to target PS-mediated immune suppression by decreasing Xkr8 expression via RNA interference (RNAi). We have developed a nanoparticle (NP) with surface coating of chondroitin sulfate (CS) and PEG-CS that can selectively co-deliver Xkr8 short interfering RNA (siXkr8) and a prodrug conjugate of 5-fluorouracil (5-Fu) and oxoplatin (FuOXP) [29] to the tumors. Combination chemotherapy regimen FOLFOX including oxaliplatin, fluorouracil and folinic acid has been a major treatment for colorectal and pancreatic cancers. FuOXP was designed to achieve codelivery of 5-Fu and oxoplatin, and was expected to release Pt (II) and 5-FU upon intracellular delivery. A series of similar “dual-prodrug” conjugates have been reported in which the hydroxyl group of oxoplatin was linked to the N-1 of 5-FU for cooperative cytotoxicity against cancer cells [29]. Our NP delivery platform works through effective tumor endothelial cells (ECs)-mediated active targeting while minimizing liver sinusoidal ECs (LSECs)-mediated liver uptake. Our recent work highlighted the pivotal role of tumor endothelial cells' CD44-mediated internalization and transcytosis in facilitating the extravasation at the tumor site. Meanwhile, the tumor cells' CD44-mediated transcytosis contributes significantly to the deep tumor penetration of our NP. This strategy showed promising therapeutic efficacy as well as excellent safety profile [4].
In this study, we extended the investigation of this strategy to two subcutaneous (s.c.) pancreatic tumor models (Panc02 and KPC) with different responses to chemodrug-induced Xkr8 expression. We further studied the role of impaired efferocytosis in the TME after co-treatment. Finally, we co-delivered FuOXP and siXkr8 in a clinically relevant orthotopic pancreatic cancer model for evaluating tumor inhibition and anti-tumor immunity.

留言 (0)