
Remember me
Multiple myeloma (MM) is a hematological malignancy associated with the proliferation of plasma cells in the bone marrow, making it a complex and challenging disease to treat.[1] Over the years, advancements in research have led to the development of therapeutic approaches, including chemotherapy, immunomodulatory drugs, and stem cell transplantation.[2,3] However, despite these therapeutic options, MM remains incurable in most cases, necessitating the exploration of novel therapies.[4]
A promising and emerging treatment approach with significant potential for patients with MM is the anti-B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T-cell therapy.[5,6] The therapy revolves around genetically modifying the patient’s T-cells to express CARs, allowing them to identify and eradicate myeloma cells[5] efficiently. The ability to target BCMA lies in its high expression on the surface of malignant plasma cells, making it suitable for CAR T-cell therapy in MM.[6] The rationale behind targeting BCMA lies in its selective expression on myeloma cells while sparing healthy tissues, making it a therapeutic target.[7] Moreover, BCMA has a crucial role in the survival and proliferation of MM cells.[7] Modifying T-cells to express CARs specific to BCMA allows researchers to understand the immune system’s capacity to eradicate myeloma cells while minimizing off-target toxicity.
Early clinical trials evaluating anti-BCMA CAR T-cell therapy for MM have yielded consistent results[7,8] in examining various CAR designs. These designs encompass second and third generations, incorporating distinct signaling domains and T-cell functionalities. The therapy’s effectiveness has been observed in refractory MM compared to traditional treatments with limited efficacy and outcomes.[9] D’Agostino and Raje[9] reported positive anti-BCMA CAR T-cell therapy outcomes. They showed that patients experienced significant improvements, including higher response rates, extended remission periods, and enhanced overall survival. Similarly, Anderson[10] evaluated idecabtagene vicleucel, a BCMA-targeting CAR T-cell therapy in MM. Their outcomes revealed a promising overall response rate of 73%, with 33% of patients attaining complete response.[10] These findings strongly indicate the potential efficacy of anti-BCMA CAR T-cell, positioning it as a viable and promising treatment option for MM.
According to Chekol Abebe et al.,[11] anti-BCMA CAR T-cell therapy has demonstrated manageable safety profiles in clinical trials.[11] Cytokine release syndrome (CRS) and neurotoxicity can be effectively managed with appropriate supportive care measures.[12] However, researchers continue to refine the therapy’s administration and dosing strategies to minimize toxicity while maximizing its anti-tumor effects. Nam et al.[13] suggested that despite these promising results, challenges remain in adopting anti-BCMA CAR T-cell therapy for MM. The manufacturing process of CAR T-cell therapies is complex and time-consuming, making it logistically demanding and costly.[13] In addition, the durability of responses and long-term safety data require further investigation to establish the therapy’s role in treating MM. To address these challenges, ongoing research is dedicated to enhancing the efficacy and safety of anti-BCMA CAR T-cell, focusing on optimizing the accessibility and affordability of the therapy. This involves streamlining manufacturing processes, reducing turnaround times, and ensuring cost-effectiveness.[13] In addition, synergistic approaches are explored by combining anti-BCMA CAR T-cell therapy with other treatments like immunomodulatory drugs or monoclonal antibodies to amplify therapeutic outcomes and broaden the therapy’s safety and efficacy.[2,3]
Our main objective is to perform a systematic review and meta-analysis to explore the efficacy and safety of anti-BCMA CAR T-cell therapy for MM.
MATERIAL AND METHODS Information sourcesOur study was performed according to the preferred reporting items for systematic reviews and meta-analysis (PRISMA) guidelines[14] and the Cochrane Handbook for systematic reviews and interventions.[15] We searched five databases: Web of Science (https://clarivate.com/products/scientific-and-academic-research/research-discovery-andworkflow-solutions/webofscience-platform/), CNKI (https://en.cnki.com.cn/index/), EMBASE (https://www.embase.com/landing?status=gray), PubMed (https://pubmed.ncbi.nlm.nih.gov/), and Cochrane (https://www.cochranelibrary.com/), for studies published on anti-BCMA CAR-T-cell treatment for MM. The databases were searched from their inception to 2023 using the following keywords; “Myeloma cells,” “CAR,” and “BCMA.”
Eligibility criteriaOur study adopted the participants, intervention, comparison, and outcomes approach to determine eligible studies for inclusion. Moreover, the following inclusion and exclusion criteria were adopted;
Inclusion criteriaThe inclusion criteria encompassed prospective single-arm studies conducted in single- or multi-center settings involving patients exclusively diagnosed with MM, including refractory cases. These studies were required to report on the intervention of anti-BCMA CAR T-cell treatment for MM and provide outcomes related to the efficacy, safety, or both aspects of this treatment.
Exclusion criteriaThe exclusion criteria involved studies that combined anti-BCMA CAR T-cell treatment with other regimens and observational studies, conference papers, or reviews. In addition, studies reporting outcomes unrelated to anti-BCMA CAR T-cell treatment parameters were excluded. Finally, studies not published in English were also excluded from the study.
Data extraction and selectionOur study adopted two independent reviewers who conducted the study selection and data extraction. These reviewers scrutinized the selected databases using the keywords and search terms. The articles were retrieved by screening their titles and abstracts. Furthermore, the full texts were screened to ensure sufficient information available based on the study population, assessment of outcomes, and other confounding factors. A third reviewer and majority vote resolved any conflicts between the reviewers.
We obtained the following information: Year of publication, author’s surname, features of patients, features of anti-BCMA CAR-T administered, and the outcomes of safety and efficacy of the intervention. The measures of efficacy outcomes were assessed based on the rate of overall, complete, and duration of responses. Furthermore, progression-free and overall survival was determined. The rates of overall response ranged from complete to partial responses. Our safety outcomes measures were based on the grades of CRS, CAR-T encephalopathy syndromes, and other secondary infections such as anemia (Neurotoxicity).
Quality assessmentOur study adopted the Cochrane risk of bias tool (version 2.0, Oxford, England: The Cochrane Collaboration, 2003) to assess the risk of bias based on five domains; adequate random sequence generation ensures unbiased participant allocation, while allocation concealment prevents biased group assignments. Blinding of participants and personnel minimized performance and detection bias. Handling incomplete outcome data helps reduce attrition bias, and reporting all planned outcomes and analysis protects against selective reporting bias. Furthermore, we only included studies meeting the inclusion criteria and had a relatively low risk of bias based on all assessment domains. In addition, the quality of the selected publications was assessed using the Newcastle-Ottawa scale (NOS).[16] The NOS consisted of eight items that were subdivided into three main categories: Selection of cases (0–4 points), compatibility of groups (0–2 points), and clinical outcomes (0–3 points). High-quality studies are those studies that attained a score of 6 and above on the NOS scale. We found it essential to assess the risk of bias in selected publications because it minimizes false reporting and increases the accuracy of our study.
Statistical analysisOur study used Review Manager (RevMan) (version 5.4.1 RevMan (Computer program) Version 4.2 for Windows. Oxford, England: The Cochrane Collaboration, 2003) and R software (version 4.2.3, The R core team, Boston, USA) to perform all statistical analyses. We obtained forest plots, publication bias plots, and risk of bias plots. We extracted data on the response rates and incidences of adverse outcomes to perform comparisons and subgroup analyses. Furthermore, all the analyses were produced with their corresponding 95% confidence intervals (CIs). Heterogeneity was assessed using the Higgins I^2 statistic and Chi-square statistic. Heterogeneity was inferred at an I^2 ≥ 50% with P < 0.1. In such cases, the random effects model was used for analysis. Furthermore, we carried out a subgroup analysis to examine sources of heterogeneity and a sensitivity analysis with the heterogeneous studies being omitted. Our subgroup analyses were conducted based on the overall response, complete response, grades of CRS, antigens targeted by CAR, and neurologic toxicities.
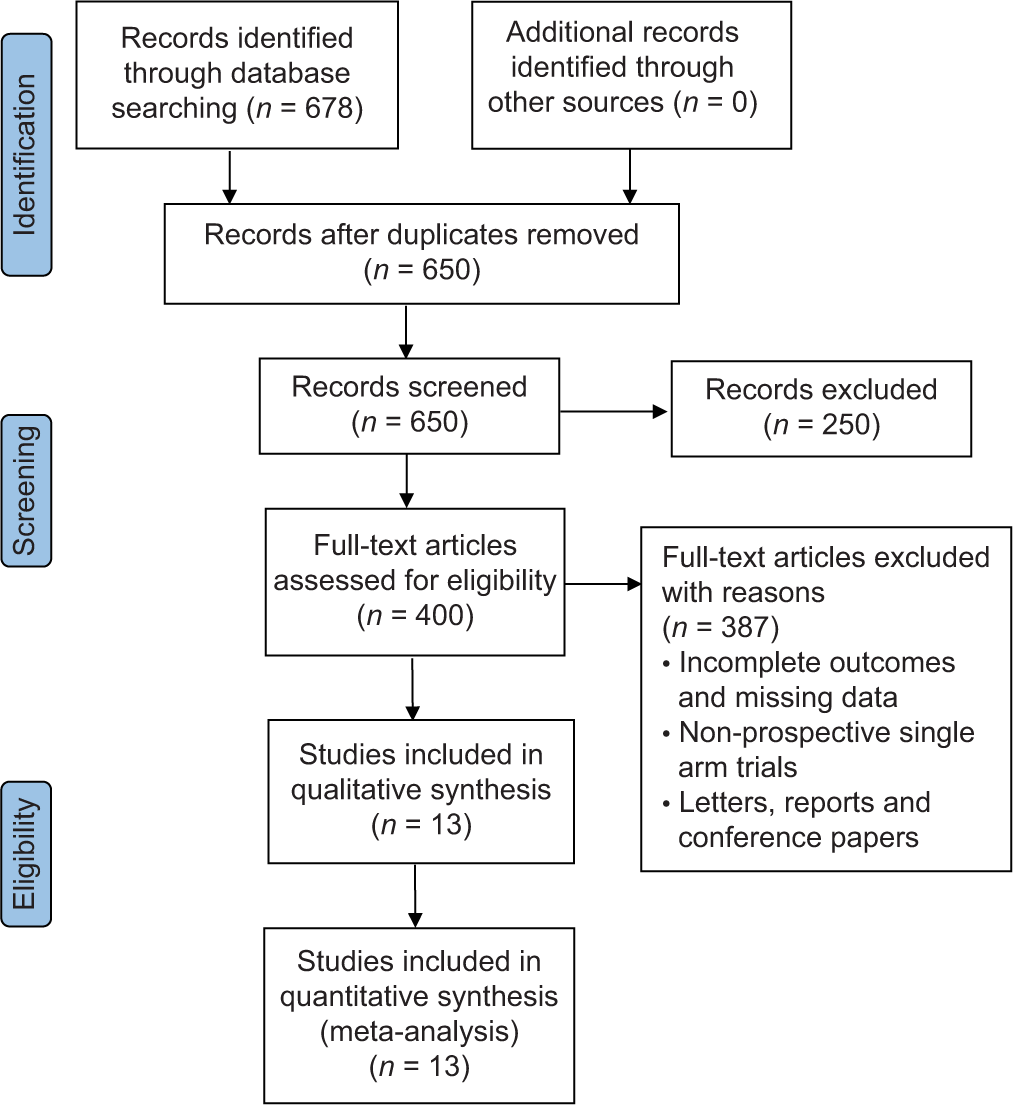
RESULTS Search resultsThe literature search produced 678 studies, of which 650 were screened based on their titles and abstracts. After applying the eligibility criteria, 400 full-text articles were reviewed, with 387 full-text articles excluded. Finally, 13 articles were eligible for analysis [Figure 1 and Table 1].
Export to PPT
Table 1: Characteristics of included studies.
Author’s surname Year of publication No. of Patients Age/gender Prior treatment, ASCT, and lymphodepletion Antigen positivity, the origin of T-cells, and subset of T-cells CAR: Vector/co-stimulatory molecule/scFvspecies CAR-T dose and follow-up Brudno et al.[17] 2018 26 Age: 18–70 years.We included studies published from 2016 to 2022 and were single-arm studies.[17-29] The participants were middle-aged, about 40 years and had an average oncology performance. In all the studies, the patients were diagnosed with MM or cases of refractory MM. We observed that patients were subjected to prior treatments using proteasome inhibitors, autologous stem cell transplantation (ASCT), or immunotherapy regimens. In most studies, patients were subjected to ASCT before treatment, while some studies did not report on the outcomes of ASCT. Lymphodepletion was mainly carried out using melphalan, cyclophosphamide, and fludarabine. However, some studies did not use this technique, and others reported incomplete outcomes.
In these studies, the extracted T-cells underwent genetic modifications to express levels of CAR in the presence of retroviruses or transposons. Most T-cells were autologous, with a single study reporting from both autologous and autologous T-cells.
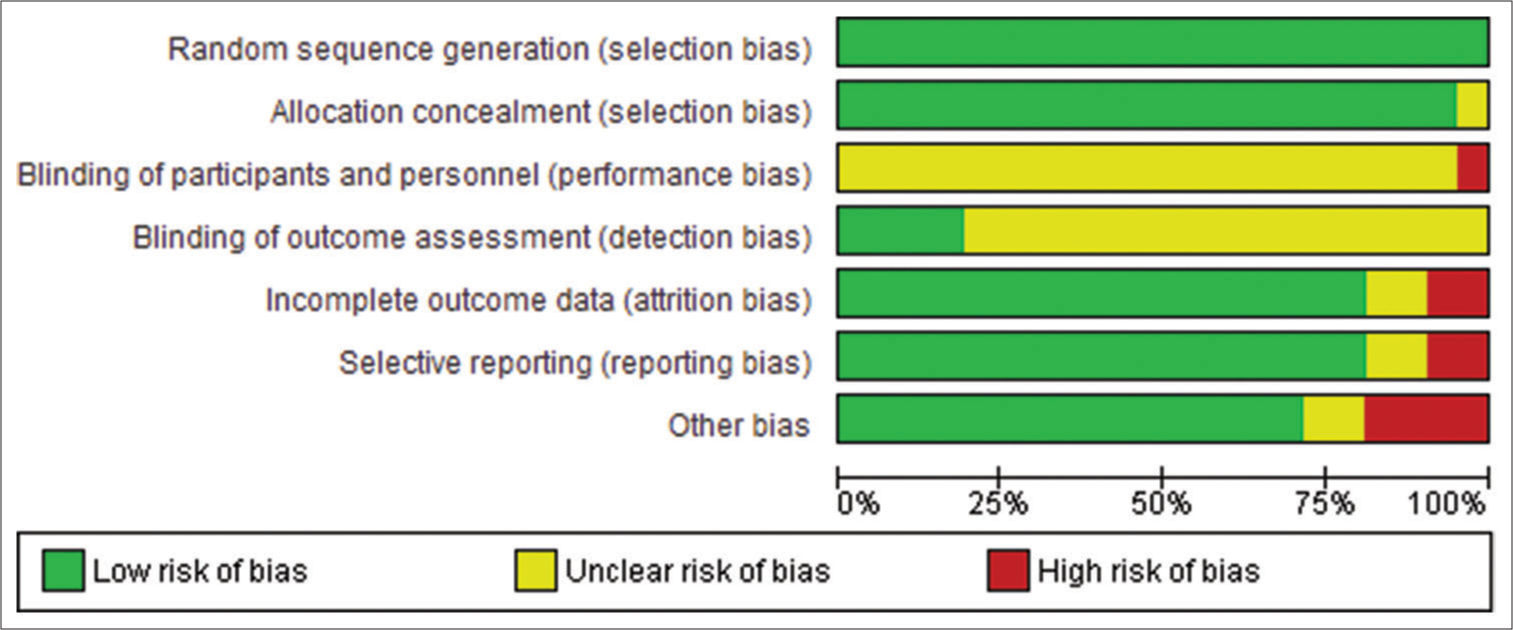
Risk of biasAccording to Figure 2, most studies had a low risk of bias in all five domains. There was a high unclear risk of bias in blinding participants and personnel because it was impossible to blind the medical professional and personnel who offered the anti-BCMA CAR-T treatment regimen to the participants. Blinding reduces the risk of bias by minimizing the influence of participant’s and personnel’s expectations or preferences on the study outcomes. Lack of blinding can introduce performance or detection bias, affecting the trial’s internal validity. Double-blind designs are ideal, where participants and investigators are unaware of the assigned interventions. Moreover, some studies reported incomplete outcomes, such as the duration of follow-up and the number of participants in the experimental and control conditions. Nonetheless, we observed moderate to high methodological qualities of all selected publications based on the ratings of the NOS Scale. Despite some incomplete outcomes, all the included studies had sufficient information for meta-analysis.
Export to PPT
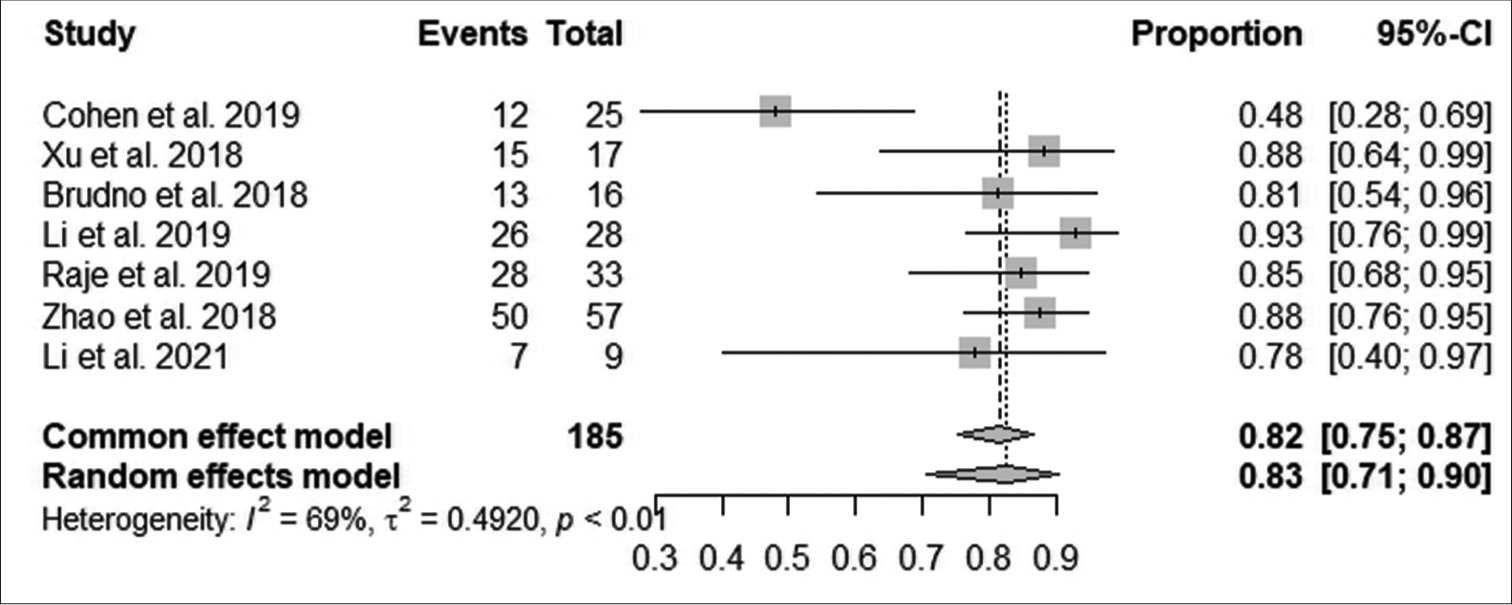
Analysis of outcomesAccording to Figure 3, the pooled proportion of the overall response rate was 82% observed from 185 patients in seven studies, with a median response within 10 months in 151 patients. The increase in overall response was statistically significant (95% CI: 75–87; I^2 = 69%; P < 0.001).
Export to PPT
According to Figure 4, the pooled proportion of complete response rate was 45% observed from 185 patients in 7 studies, with 83 patients having complete responses. The increase in complete response was statistically significant (95% CI: 38–52; I^2 = 81%; P < 0.001).

Export to PPT
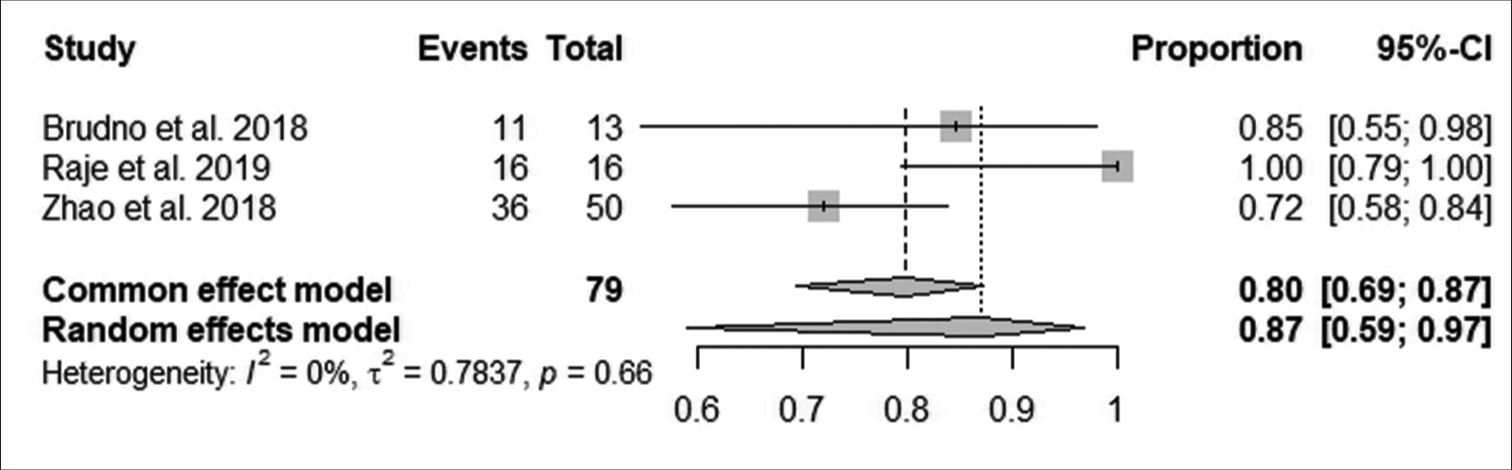
According to Figure 5, the pooled proportion of reduction in minimal residual disease negativity (MRDN) was 80% observed from 79 patients in three studies, with 63 patients having reductions in MRDN. The reduction in MRDN was not statistically significant (95% CI: 69–87; I^2 = 0%; P > 0.05).
Export to PPT
According to Figure 6, the pooled proportion of overall survival rate after follow-up was 83% observed from 148 patients in five studies, with 123 improving after 11 months. The increase in overall survival after follow-up was statistically significant (95% CI: 76–88; I^2 = 80%; P < 0.001).

Export to PPT
According to Figure 7, the pooled proportion of CRS grades 3 and 4 after follow-up was 17% observed from 185 patients in seven studies, with 31 having CRS grades 3 and 4. CRS grade 3 and 4 reduction after follow-up was statistically significant (95% CI: 12–23; I^2 = 68%; P < 0.05).

Export to PPT
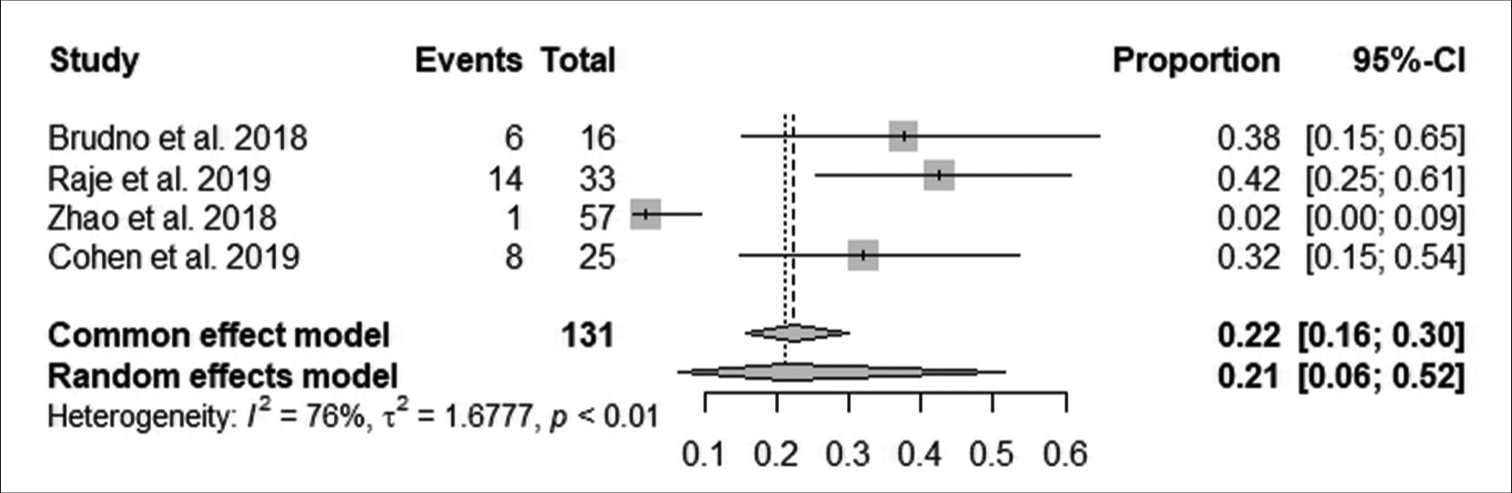
According to Figure 8, the pooled proportion of neurotoxicity after follow-up was 22% observed from 131 patients in four studies, with 29 having neurotoxicity. The reduction in neurotoxicity after follow-up was statistically significant (95% CI: 16–30; I^2 = 76%; P < 0.05).
Export to PPT
DISCUSSIONCAR-T therapy has revolutionized cancer treatment, offering a novel approach to addressing malignancies like MM. This therapy has shown remarkable promise in improving clinical outcomes by genetically modifying patients’ T-cells to express CARs targeting specific antigens on cancer cells.
Our study comprehensively evaluated anti-BCMA CAR T-cell therapy’s efficacy by examining overall response rates, complete response rates, MRDN, and overall survival. The outcomes revealed a statistically significant improvement in overall, complete, and survival response rates. However, the reduction in MRDN was not significant. The reduction in MRDN underscores the therapy’s ability to control the disease robustly. Moreover, the observed reduction in neurotoxicity further accentuates the promising potential of anti-BCMA CAR T-cell therapy in MM.[30] Anti-BCMA CAR T-cell therapy enhanced the overall response rate in MM through a series of interconnected mechanisms. These CAR T-cells are initially designed to precisely recognize BCMA, highly expressed on malignant plasma cells in MM. This specificity ensures that CAR T-cells selectively target MM cells while sparing healthy tissue, minimizing collateral damage.[30]
The mechanism of anti-BCMA CAR T-cell therapy lies in the design of CARs. These synthetic receptors consist of an extracellular domain designed to recognize BCMA expressed on malignant plasma cells in MM. The binding of CAR T-cells to BCMA initiates a cascade of intracellular signaling events, leading to the activation of cytotoxic molecules such as perforin, granzymes, interferon-gamma, and tumor necrosis factor, contributing to a robust immune response against MM cells.[31] Perforin plays a critical role by forming pores in the cell membrane of the MM cells, creating channels for granzymes to enter. Once inside the target MM cells, granzymes trigger biochemical events, leading to apoptosis or programmed cell death.[31] This apoptosis induction is a fundamental mechanism by which anti-BCMA CAR T-cells effectively eliminate MM cells.
Our study’s results aligned with Cornell et al.,[32] who outlined the genetic modification process in anti-BCMA CAR T-cell therapy. The improvement in complete and overall response rates is attributed to its persistence in the patient’s body. The ability of CAR T-cells to recognize BCMA-expressing myeloma cells increases its efficacy as a therapeutic target in MM.[31] Incorporating CD28 signaling domains in CAR design enhances T-cell activation and persistence, with cytokine production sustaining the anti-tumor immune response.[33-37] These results reaffirm anti-BCMA CAR T-cell therapy’s potential to advance MM treatment[31] significantly.
The strategic inclusion of CD28 signaling domains within CAR design amplifies the activation and persistence of T-cells. This approach stimulates cytokine production, sustaining the anti-tumor immune response, and fostering improved overall patient survival.[37] Honikel and Olejniczak[36] showed that illuminating the significance of CD28 allows an understanding of its role as a potent costimulatory signal for T-cell activation and function. The interactions between CAR design and T-cell biology outline the therapy’s multifaceted effectiveness in combating MM. The presence of CD28 signaling domains within the CAR design significantly enhances the activation and persistence of CAR T-cells. This heightened activation leads to a more effective immune response against MM cells. As a result, patients treated with anti-BCMA CAR T-cells containing CD28 signaling domains experience improved overall responses, with a more significant proportion of MM cells being targeted and eliminated.[37,38]
The effective management of neurotoxicity is a critical concern in CAR T-cell therapy, particularly the emergence of CAR T-cell-related encephalopathy syndrome (CRES). Our findings are similar to Kotch et al.,[39] who advocate for a methodical approach encompassing accurate dose optimization and a comprehensive evaluation of risk factors. The multifaceted strategy is pivotal in mitigating the potential impact of CRES. Central to this approach is the concept of patient-centric customization, where a tailored administration of CAR T-cell doses coupled with a deep understanding of individual patient attributes and vigilant response monitoring synergistically contribute to a substantial reduction in both the likelihood and severity of neurotoxicity. Anti-BCMA CAR T-cells are designed to recognize and target MM cells expressing BCMA. This targeted approach ensures that CAR T-cells selectively attack MM cells, particularly those that may be resistant to conventional therapies. As a result, the number of remaining MM cells is reduced, contributing to MRD negativity. Furthermore, the therapy is designed to minimize off-target effects on healthy tissues. This design reduces the risk of CRES that can occur when CAR T-cells affect non-tumor brain tissue. Thus, by focusing their activity primarily on BCMA-expressing MM cells, anti-BCMA CAR T-cells aim to spare healthy brain tissue, thus lowering the incidence and severity of CRES.
Dose optimization is crucial in precision medicine, ensuring proper alignment of therapeutic intervention with individual patient needs. Balancing efficacy and minimizing adverse effects lowers neurotoxicity risks and enhances tolerability during treatment. This approach advances personalized medicine, reflecting a profound understanding of CAR T-cell therapy.[39] Interventions such as tocilizumab, an interleukin-6 receptor antagonist, emerge as potential factors in minimizing adverse effects. Tocilizumab’s mechanism is associated with immune response and targeting the interleukin-6 receptor. Tocilizumab intercepts a critical signaling pathway in CRS development with a positive outcome in improved immune response modulation and dampening the intensity of CRS.[40]
The diverse strategies for managing neurotoxicity and CRS are critical for cancer treatment. Balancing therapeutic efficacy and patient well-being is central to effective and safe treatment. In CAR T-cell therapy, proactive measures such as precise dosing and targeted tocilizumab protect against adverse effects. Thus, by carefully administering these strategies, Healthcare professionals optimize the therapy while prioritizing patient safety. The previous studies[41-44] showed that anti-BCMA CAR T-cell therapy demonstrated a marked reduction in MRDN, primarily achieved through the targeted elimination of malignant plasma cells. This approach ensures the direct and efficient killing of myeloma cells, even those resistant to conventional therapies.[42] The persistence of CAR T-cells and continuous targeting of residual myeloma cells contributes to sustained disease control, minimizing the risk of relapse.[40-42]
Strengths and limitationsOur study was robust because it was performed based on the Cochrane guidelines and PRISMA protocols. We used an extensive search strategy and the NOS scale to assess the methodological quality of selected publications. Nonetheless, our study was limited by heterogeneous data of the selected publications and variations in the doses administered. In addition, some of the studies had significant variations in the duration of follow-up. Further studies should adopt randomized controlled trials involving experimental and control groups in MM.
SUMMARYOur study suggested that using anti-BCMA CAR T-cell therapy in MM has high efficacy and safety in mitigating adverse outcomes such as MRDN, neurotoxicity, and CRS. In addition, this therapy improved survival outcomes, complete response rates, and overall response rates in MM patients. We found that the extracellular domain of CARs allows the CAR T-cells to recognize and bind specifically to BCMA on the surface of myeloma cells. Once infused back into the patient’s body, the modified CAR T-cells circulate and encounter BCMA-expressing myeloma cells.
AVAILABILITY OF DATA AND MATERIALSThe data that support the findings of this study are available within the article or its supplementary materials.
ABBREVIATIONSMM - Multiple Myeloma
BCMA: B-cell maturation antigen
CD28: Cluster of Differentiation 28
CD14: Cluster of Differentiation 14
ASCT: Autologous stem cell transplantation.
CAR-T: Chimeric antigen receptor T-cell therapy
Flu/Cy: Fludarabine and cyclophosphamide.
4-1BB (CD137): Cluster of Differentiation 137
AUTHOR CONTRIBUTIONSJZ, XD and XD designed the research study, performed the research. XD and XD provided help and advice on the experiments. XD analyzed the data. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Comments (0)