
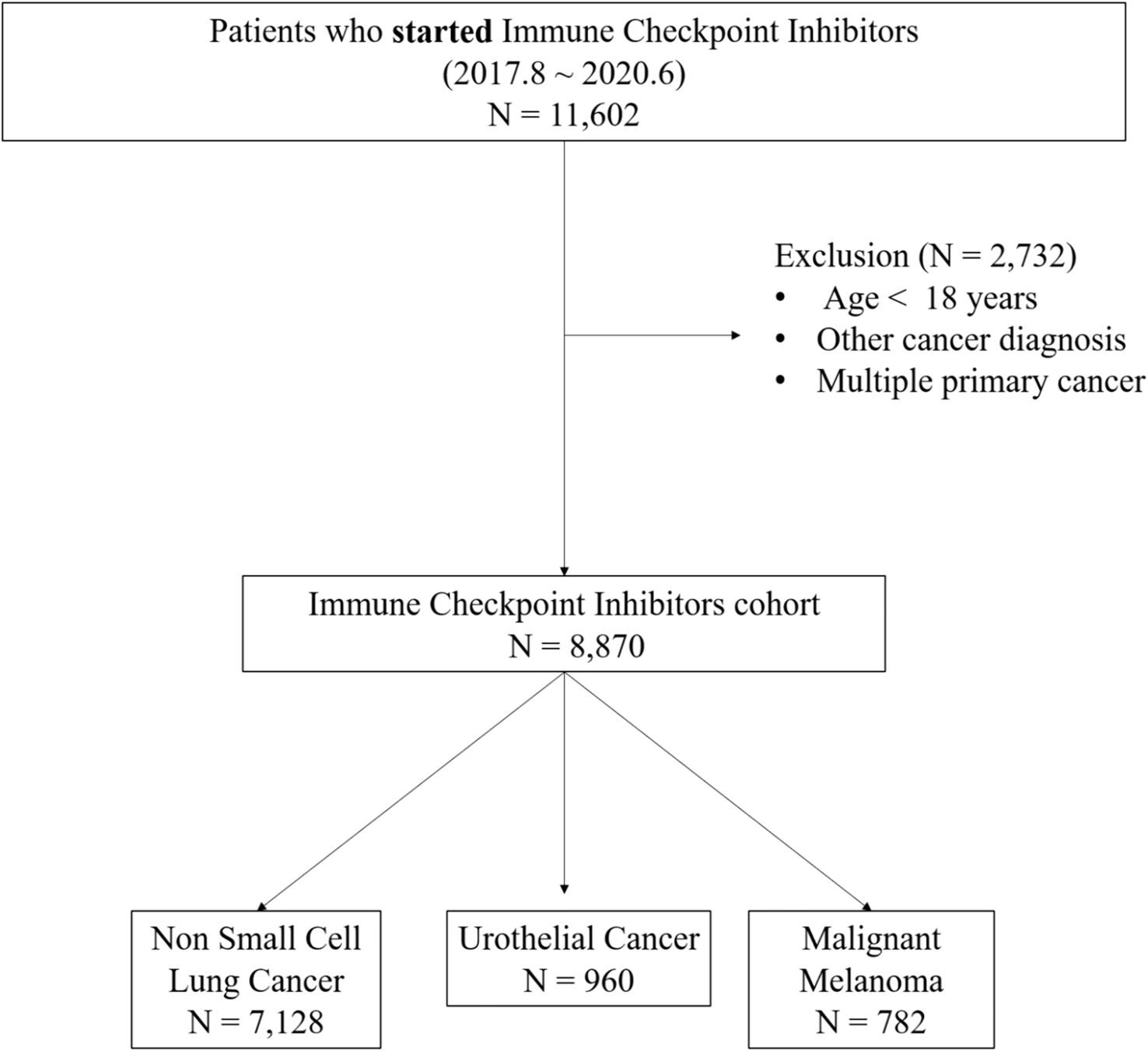
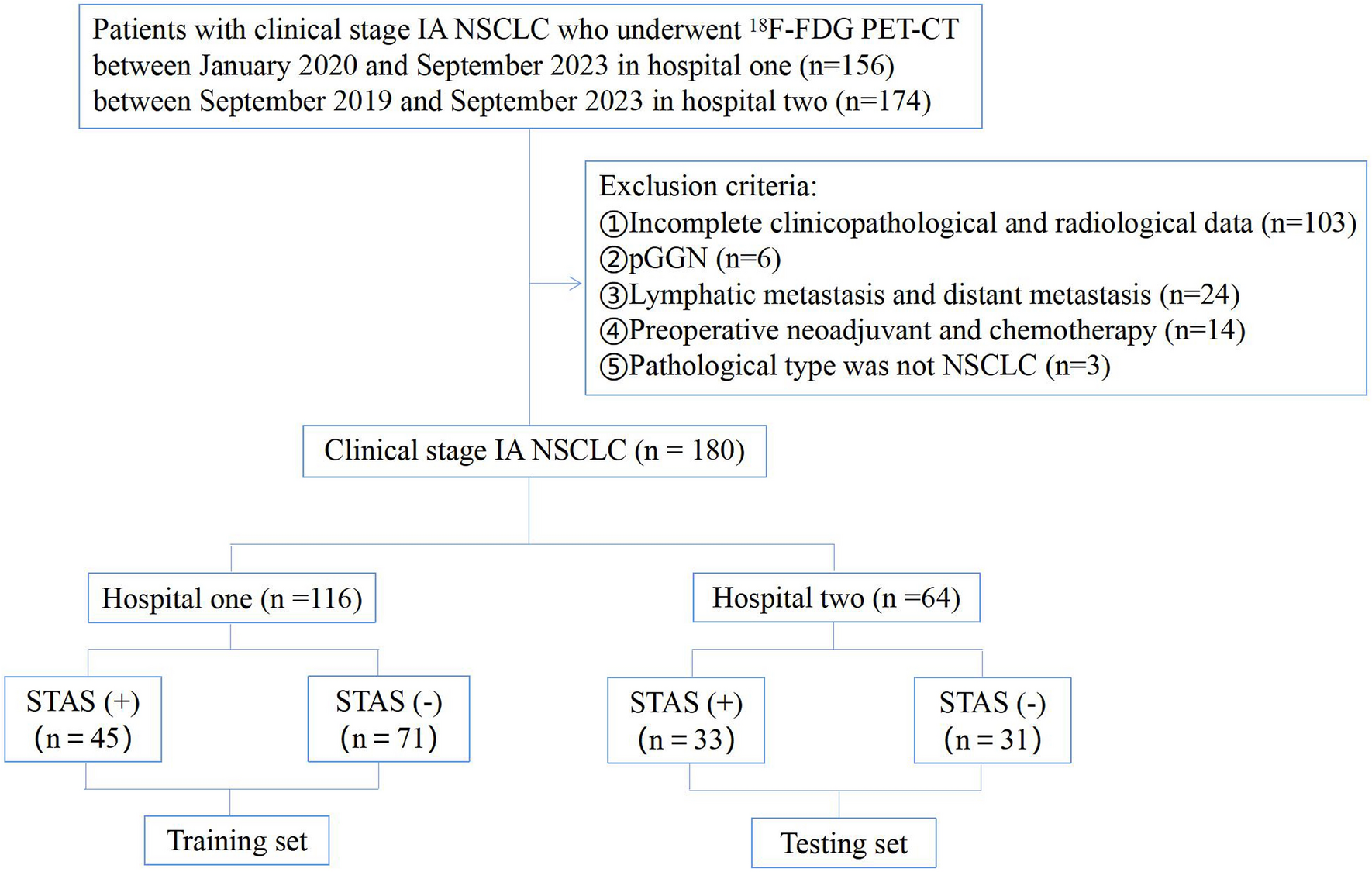
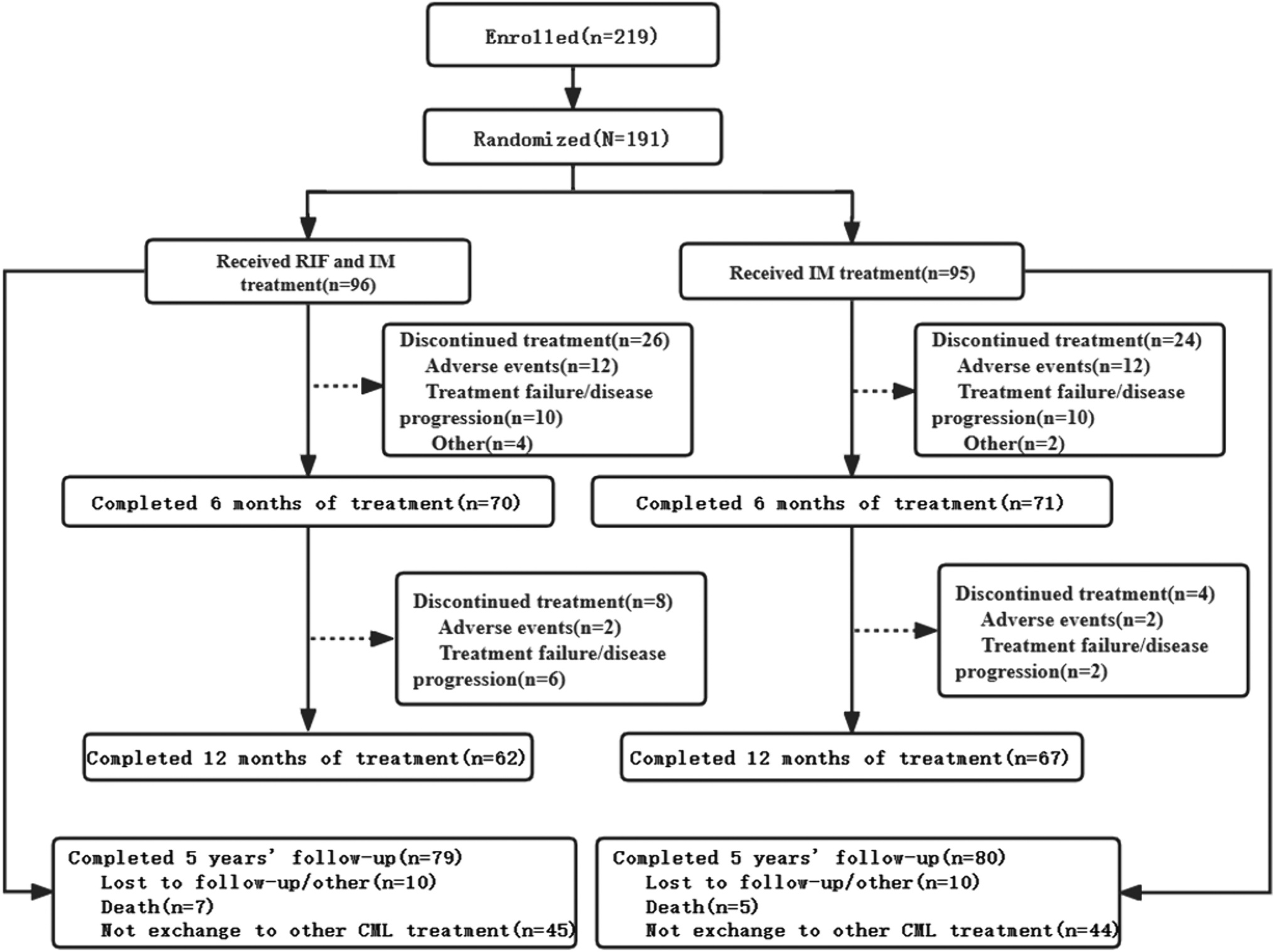
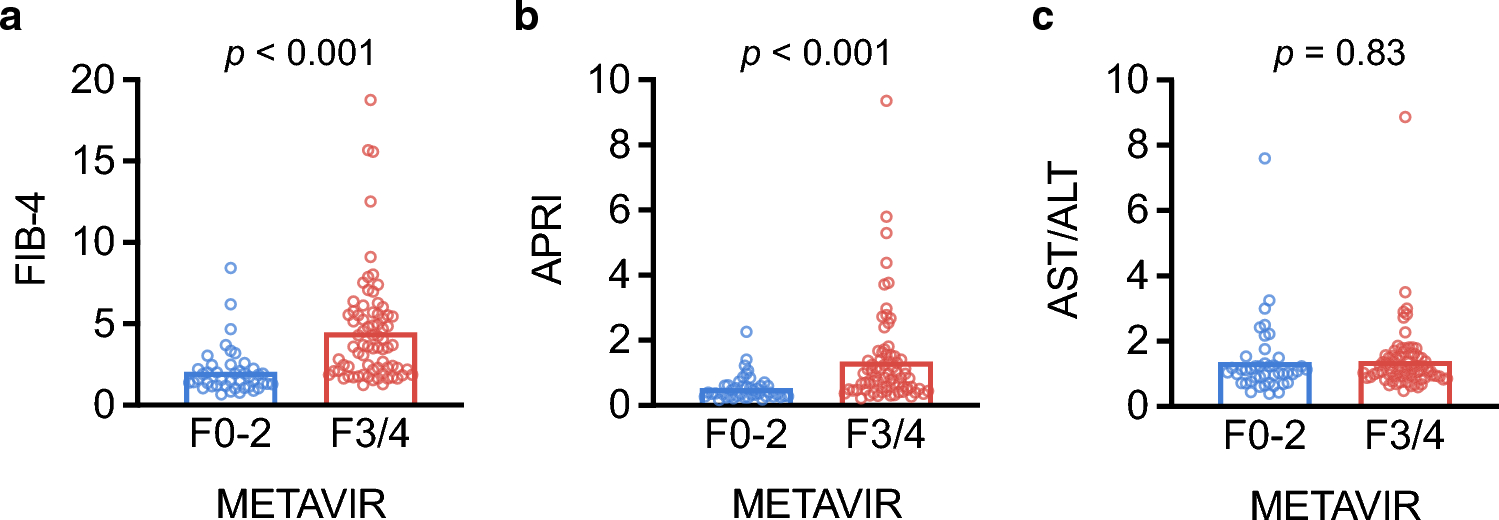
記住我
The general work flow of our study is depicted in Fig. 1. A total of 911 normal soft tissue samples (all from GTEx) and 259 STS patients (all from TCGA) were enrolled in this study. By combining the data from GTEx and TCGA, we collected 4652 lncRNAs. The expression data patterns of 67 NRGs from the GSEA set and relevant literature were acquired, and 454 necroptosis-related lncRNAs was finally extracted using Pearson correlation analysis (|coefficient| > 0.4 and p < 0.001). The correlation between these necroptosis-related lncRNAs and corresponding NRGs observed in networks suggests they have an intricate relationship (Fig. S1A). Furthermore, we identified 39 prognosis-specific necroptosis-related lncRNAs with STS via univariate Cox regression analysis (p < 0.02) and presented in the forest plot (Fig. S1B). Among these, the expressions of 22 lncRNAs were significantly higher, whereas 17 lncRNAs were lower in tumors (p < 0.001; Fig. S1C). Taken together, our study indicated that these prognostic necroptosis-related lncRNAs might play a pivotal role in the progression of STS.
Fig. 1
Flow diagram of this study
Unsupervised consensus clustering analysisSubclusters in STS identified by unsupervised consensus clustering were reported to have different TME and immunotherapy response (Das et al. 2020; DeBerardinis 2020). K = 2 was confirmed as the optimal number of clusters (Fig. 2A). Therefore, hierarchical unsupervised clustering of the 39 necroptosis-related lncRNAs revealed two major clusters (i.e., cluster 1 and cluster 2). Survival analysis evidenced that STS patients in the cluster 2 had a worse OS than those in cluster 1 (Fig. 2B), and a significant differences in age, histological type, margin status and radiotherapy were observed in the heatmap between cluster 1 and cluster 2, indicating the potential correlations between clinical characteristics and 39 necroptosis-related lncRNAs (Fig. 2C). The abundance of eight immune cells, including resting T cells CD4 memory, activated T cells CD4 memory, M0 macrophages, M2 macrophages, resting Dendritic cells, resting Mast cells, Eosinophils and Neutrophils, were significant difference in cluster 1 and cluster 2 (Fig. S2A). The abundance of activated T cells CD4 memory, M0 macrophages, M2 macrophages, Eosinophils and Neutrophils was significantly higher, whereas that of resting T cells CD4 memory, resting Dendritic cells and resting Mast cells was lower in cluster 2 than in cluster 1 (Fig. S2B). This result indicated the potential connection between immune infiltrate cells and 39 necroptosis-related lncRNAs. Meanwhile, the expression levels of several immune checkpoints, including, PD-L1, BTNL2 and VTCN1, were markedly higher in cluster 2 than in cluster 1, whereas that of PD-1 and CTLA4 was lower in cluster 2 than in cluster 1 (Fig. 3A–C; Fig. S3). The correlations between these immune checkpoints and 39 necroptosis-related lncRNA were displayed in Fig. 3D–F. Summing up our observations, the finding of our data showed a significant correlation between necroptosis-related lncRNAs and the prognosis as well tumor-immune landscape in STS.
Fig. 2
Consensus clustering based on necroptosis-related lncRNAs in STS. A Consensus clustering matrix for k = 2. B Kaplan–Meier analysis of patients in cluster 1 and cluster 2 subgroups. C Heatmap and clinicopathologic features of the two clusters (* p < 0.05; ** p < 0.01; *** p < 0.001)
Fig. 3
Necroptosis-related lncRNAs were correlated with immune checkpoints in STS. The expression of immune checkpoints A PD-L1, B BTNL2 and C VTCN1 in cluster 1 and cluster 2 subgroups. D–F Co-expression analysis of immune checkpoints and 39 necroptosis-related lncRNAs
Gene set enrichment analysis in subclustersWe further performed GSEA analysis between the subclusters to investigate potential functions of 39 necroptosis-related lncRNAs. The finding of our data illustrated that multiple signaling pathways, including KEGG_APOPTOSIS, KEGG_N_GLYCAN_BIOSYNTHESIS, KEGG_PANCREATIC_CANCER, KEGG_WNT_SIGNALING_PATHWAY, and KEGG_NOD_LIKE_RECEPTOR_SIGNALING_PATHWAY, were remarkably enriched in cluster2, and KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION, KEGG_PROPANOATE_METABOLISM, KEGG_CALCIUM_SIGNALING_PATHWAY, KEGG_INSULIN_SIGNALING_PATHWAY and KEGG_FATTY_ACID_METABOLISM were more enriched in cluster 1 (Fig. S4). These results provided important insights into the potential cross-signaling pathways in STS, which modulated by necroptosis-related lncRNAs.
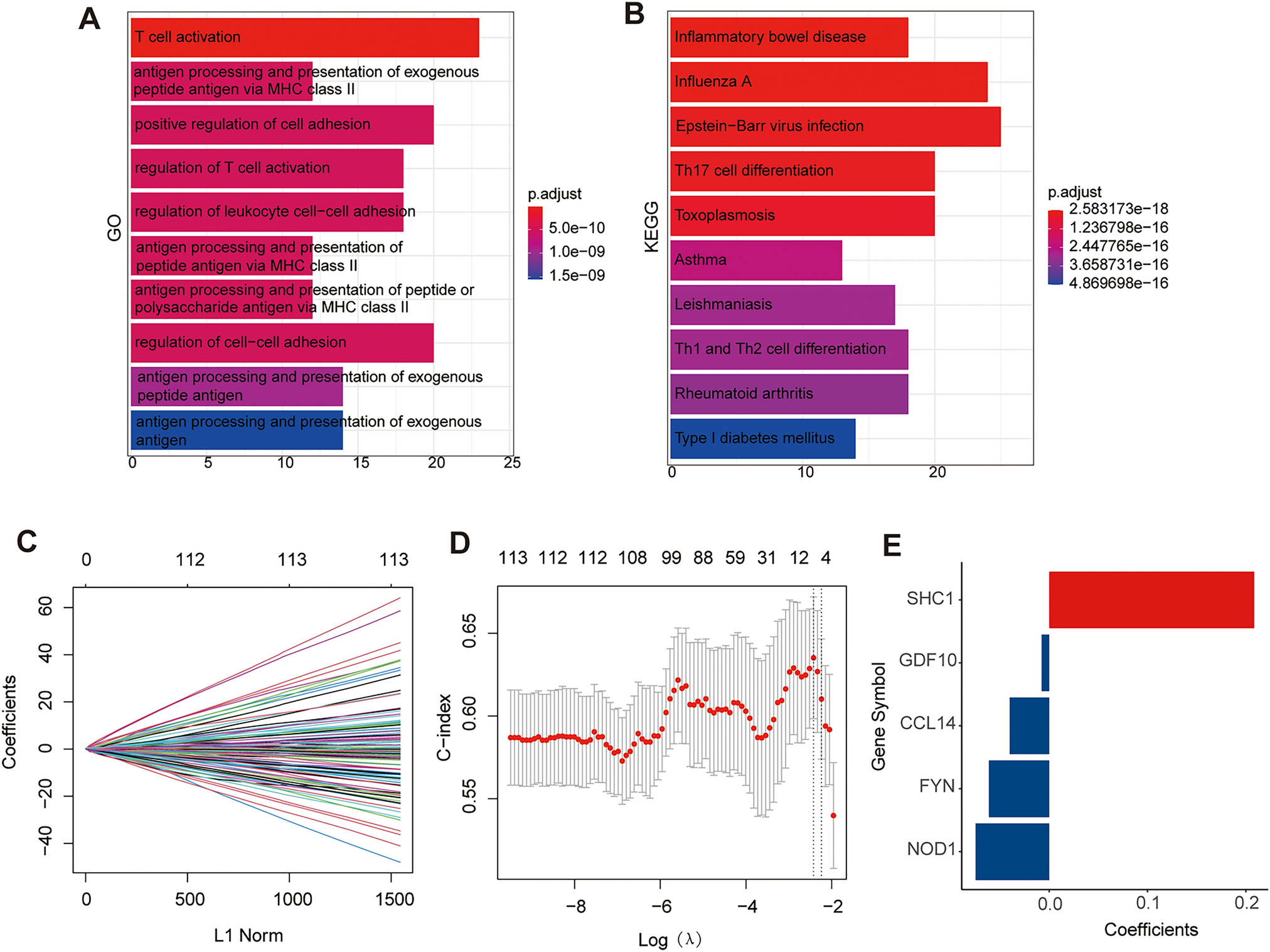
Construction and verification of the risk signatureWe finally identified the eight-hub necroptosis-related lncRNAs based on the result of Lasso Cox regression analysis to prevent overfitting (Fig. 4A, B). The relationship between these eight lncRNAs and corresponding NRGs is shown in Fig. 4C. The comparison in the expression of these eight lncRNAs expression suggested that the expressions of LINC02454, SERTAD4-AS1, POLH-AS1 and HEATR6-DT were notably higher, whereas IRF1-AS1, SNHG1, IGF2-AS and SLC9A3-AS1 were more lowly expressed in tumor samples in comparison with normal soft tissue samples (p < 0.001; Fig. 4D). Furthermore, the risk score of every STS patient is calculated in the formula as follows:
$$}\,} = }02454\,}. \times (0.0491) + }1 - }1\,}. \times ( - 0.3121) + }4 - }1\,}. \times ( - 0.1332) + } - }1\,}. \times (0.3079) + }6 - }\,}. \times (0.2914) + }1\,}. \times (0.0858) + }2 - }\,}. \times (0.0572) + }9\,}3 - }1\,}. \times (0.3491)$$
Fig. 4
Construction of NRLncSig in STS. A,B LASSO analysis with minimal lambda value. C The relationship between the hub prognostic necroptosis-related lncRNAs and corresponding NRGs. Blue: NRGs. Red: lncRNAs. D The boxplot of the hub prognostic necroptosis-related lncRNAs in both tumor and normal samples. NRGs, necroptosis-related genes; NRLncSig, necroptosis-related lncRNA-based risk signature (* p < 0.05; ** p < 0.01; *** p < 0.001)
We defined this risk signature as NRLncSig. The prognostic performances of the NRLncSig were evaluated using external validation, including the test set (n = 128) and entire set (n = 259). The STS patients could be clearly divided into low- and high-risk groups in each set based on the medium risk score. The survival analysis suggested that high-risk group had a worse prognosis whether in the train, test, or entire sets (Fig. 5A–C). Time-dependent receiver operating characteristics (ROC) curve illustrated that the area under the ROC curve (AUC) of 5-year AUC in the train set, test set and entire set was 0.767, 0.690, and 0.735, respectively (Fig. 5D–F). Besides, with the risk score formula, the distribution of risk score, the survival status, survival rate, and the corresponding expression data of these hub lncRNAs of patients were compared between low- and high-risk groups in the train, test, and entire sets (Fig. 5G–I). The results all evidenced that the high-risk group had the worse OS.
Fig. 5
Prognosis value of the NRLncSig in the train, test, and entire sets. A–C Kaplan–Meier survival curves of overall survival of patients between low- and high-risk groups in the train, test, and entire sets, respectively. D–F The 1-, 3-, and 5-year ROC curves of the train, test, and entire sets, respectively. G–I Survival time, survival status and the hub lncRNAs expression between low- and high-risk groups in the train, test, and entire sets, respectively. NRLncSig, necroptosis-related lncRNA-based risk signature
Clinical evaluation of the NRLncSigWe performed survival analyses on the NRLncSig of patients with different age, sex, recurrence, metastasis, radiation therapy, margin status, and histological type to further verify the prognostic value of the NRLncSig in various STS patients. High-risk group was confirmed to have a worse OS for both male and female patients, regardless of whether they were ≤60 or >60 years old, with or without metastasis, with positive margin status or negative margin status, with or without recurrence, and whether they were patients with DDLPS and LMS (Fig. S5A–M). These results above again verified the promising prognostic predictor of the NRLncSig for STS patients, regardless of other clinical features. The relationship between the NRLncSig and clinical features was also investigated and the results showed that the NRLncSig score was observed to be significantly connected with recurrence, histological type, age, immune score, and clusters (Fig. 6A). The risk score of patients in age > 60 presented higher risk scores than those in age ≤ 60; patients with high immune scores presented a lower risk score than those with low immune scores. Patients with recurrence were higher than that of patients without recurrence; patients in cluster 2, who exhibited poor OS, presented higher risk scores than those in cluster 1; and patients with SS had a remarkably higher risk scores than those with DDLPS, LMS, MFS and UPS (Fig. 6B–F). In addition, the NRLncSig had a higher AUC of 5-year comparing with other clinical features (AUC = 0.735; Fig. 6G). All the results above illustrated that the accuracy of NRLncSig was a satisfactory and reliable prognostic predictor.
Fig. 6
Clinical evaluation of NRLncSig in STS. A The heatmap of the associations between the NRLncSig risk score and clinicopathological features. The boxplots of the associations between the risk score and B age, C immune score, D confirmed recurrence, E cluster and F histological type. G A comparison of 5-year ROC curve with other clinical characteristics. NRLncSig, necroptosis-related lncRNA-based risk signature (* p < 0.05; ** p < 0.01; *** p < 0.001)
Gene set enrichment analysis based on risk subgroupsThe results of GSEA indicated that the KEGG_BASAL_CELL_CARCINOMA, KEGG_CARDIAC_MUSCLE_CONTRACTION,KEGG_HEDGEHOG_SIGNALING_PATHWAY, KEGG_OTHER_GLYCAN_DEGRADATION, and KEGG_TGF_BETA_SIGNALING_PATHWAY were the top five most enriched signaling pathways in the high-risk group, while the KEGG_CHEMOKINE_SIGNALING_PATHWAY, KEGG_CYTOKINE_RECEPTOR_INTERACTION, KEGG_HEMATOPOIETIC_CELL_LINEAGE, KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY and KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION were most enriched in the low-risk group (Fig. 7A, B; Table S2). These results also provided novel insights into the potential pathways in STS, which was regulated by eight-hub necroptosis-related lncRNAs.
Fig. 7
Gene set enrichment analysis in the high- and low-risk groups. Top five enriched pathways enriched for high- (A) and low-risk scores (B), respectively
The immune landscape based on risk subgroupsThe heatmap of immune cell infiltrates according to diverse algorithms, including CIBERSORT, CIBERSORT-ABS, MCP counter, QUANTISEQ, XCELL, EPIC and TIMER, is demonstrated in Fig. 8A. Most significant immune cells were correlated with the low-risk group on diverse algorithms displayed at the heatmap, including macrophage M2, T cell CD4+ naive, immune score at XCELL, B cell, T cell CD4+ at TIMER, T cell CD8+ at QUANTISEQ and macrophage at MCPcounter and EPIC (Table S3). As we know, the extent of immune cell infiltration between ‘hot’ and ‘cold’ tumors differs substantially. All the results indicated the low-risk group had a higher immune infiltration status and behaved as the ‘hot’ tumor. The ssGSEA analysis showed that all the 13 immune functions were notably different between low- and high-risk groups, and the T cell functions including checkpoint, cytolytic, HLA, regulation of inflammation, co-stimulation of T cell, co-inhibition of T cell, type II INF response and type II INF response were significantly higher in the low-risk than of high-risk groups (Fig. 8B).
Fig. 8
The NRLncSig was correlated with immune cell infiltrates in STS. A Heatmap for immune responses based on CIBERSORT, CIBERSORT-ABS, MCP counter, QUANTISEQ, XCELL, EPIC and TIMER algorithms among high- and low-risk groups. B ssGSEA for the association between immune cell subpopulations and related functions. C Expression of immune checkpoints including PDCD-1 (PD-1), CD274 (PD-L1) CTLA4 and BTLA, among high- and low-risk groups. NRLncSig, necroptosis-related lncRNA-based risk signature (* p < 0.05; ** p < 0.01; *** p < 0.001)
Checkpoint inhibitors are the most thoroughly investigated class of immunotherapy to date and have transformed clinical management for a number of malignancies, such as STS. Given that the importance of cancer immunotherapy with immune checkpoint inhibitors, we further investigated the difference in the expression of immune checkpoints between the high-risk and low-risk groups and the result revealed that all 30 immune checkpoints were more active in the low-risk group, including PDCD-1 (PD-1), CD274 (PD-L1), CTLA4, LAG3, and BTLA (Fig. 8C). A negative correlation was further observed between the TME score and NRLncSig score (Fig. S6A–C) and the results indicated that the low-risk group had a higher immune score, stromal score and ESTIMATE score, which signifying a different TME from the high-risk group. Moreover, the NRLncSig score was positively with M2 macrophages, M0 macrophages, Neutrophils, memory B cells and resting NK cells, while negatively associated with M1 macrophages, resting Dendritic cells, regulatory T cells (Tregs), CD8+ T cells, resting Mast cells, naïve B cells and gamma delta T cells (Fig. S6D). These finding above evidenced that the high-risk group presented the immunosuppressive microenvironment in STS, prompting further prognosis research as well providing clinical treatment guidance.
The assessment of immunotherapy response and chemosensitivity in risk subgroupsWe further investigated the potential of NRLncSig score as predictor for immunotherapy response. We divided 259 STS patients into the high- and low-TMB group based on the optimal cutoff and evaluated the prognosis in these subgroups, which revealed that low-TMB patients had a worse OS comparing with the high-TMB patients (Fig. 9A). The combination of high-TMB and low-risk score in NRLncSig had the best OS in STS via survival analysis (Fig. 9B). This prompted us to postulate a correlation of necroptosis-related lncRNA and TMB and they cumulatively contribute to prognosis of STS patients. The waterfall diagram showed that most of the mutated genes in STS patients were higher in the high NRLncSig score group (70.34%) compared with the low score group (64.10%) (Fig. 9C, D). The top five genes with the highest mutation frequencies were TP53, ATRX, TTN, MUC16 and RB1. To investigate whether the NRLncSig possess the potential to predict chemosensitivity of patients with STS, we evaluated its chemotherapeutic efficacy. We found that higher IC50 value of some chemotherapeutic agents in low-risk patients, such as Motesanib, Axitinib and A.443654, indicating that patients with low risk were more sensitive to these chemotherapeutic drugs (Fig. 9E–H). The results suggested that the low-risk group characterized by a higher immune score, had a higher IC50 of aforementioned drugs. Results above suggested the promising potential of NRLncSig to predict response immunotherapy and chemotherapy efficacy of STS patients.
Fig. 9
The NRLncSig was correlated with tumor mutation burden (TMB) and chemosensitivity in STS. A The survival between high- and low-TMB groups in the entire set was analyzed using Kaplan–Meier curves. B The combination of TMB and NRLncSig risk score helped predict the overall survival. C,D The waterfall diagram was constructed based on different NRLncSig risk groups, blue for low and red for high. E–H The correlations between the NRLncSig and chemosensitivity in STS. NRLncSig, necroptosis-related lncRNA-based risk signature
Construction and verification of the nomogramThe hazard ratio (HR) of the NRLncSig and 95% confidence interval (CI) were 1.402 and 1.284–1.530 (p < 0.001), respectively, in univariate Cox regression while 1.553 and 1.402–1.721 (p < 0.001), respectively, in multivariate Cox regression (Fig. 10A, B). Besides, age and metastasis were observed in the forest plot as independent prognostic predictors. We established the novel nomogram combined with age and metastasis for predicting the 1-, 3-, and 5-year survival of STS patients (Fig. 10C). The calibration curve of 1-, 3-, and 5-year demonstrate that the nomogram had a good accuracy for predicting OS of individual STS patients (Fig. 10D). For this reason, this nomogram is a good predictive tool and might be have the potential for clinical applications in future.
Fig. 10
A prognostic signature-based nomogram for predicting 1-, 3-, and 5-year survival in STS patients. A,B Univariate and multivariate Cox analyses of the NRLncSig risk score and clinical variables in the entire set. C A nomogram for predicting survival in the entire set. D Nomogram calibration plots for predicting overall survival at 1, 3, and 5 years in the entire set
留言 (0)