
記住我



Diabetes, a chronic metabolic disease, has seen an increasing global prevalence. Prolonged exposure of diabetic patients to high levels of glucose and fat in the environment triggers a sequence of lesions within the body’s microvasculature, termed diabetic microangiopathy (DMC). DMC is mainly categorized into diabetic kidney disease (DKD), diabetic retinopathy (DR), diabetic cardiomyopathy (DCM), and diabetic peripheral neuropathy (DPN) (Schiborn and Schulze, 2022), and primarily affects the kidney, retina, myocardium, nerve tissue, and toes. Diabetic foot complications often manifest in the advanced stages of DPN. Presently, the precise pathogenesis of DMC remains elusive in modern medicine and is likely associated with inflammatory responses, oxidative stress, vascular endothelial cell damage, and alterations in vascular permeability due to elevated glucose levels (Singh and Kulkarni, 2022; Zhu et al., 2023). Current clinical treatments for DMC primarily target these factors but exhibit limited efficacy (Nellaiappan et al., 2022). Hence, there is an urgent need to identify therapeutic targets and innovate new drugs for DMC.
Autophagy, akin to apoptosis and senescence, is a critical biological phenomenon. It degrades misfolded proteins and damaged organelles into smaller components, providing cells with necessary nutrients and materials for self-renewal (Maiuri et al., 2007). This process serves as a self-protective mechanism enabling cells to sense external stimuli or abnormal energy metabolism, crucial for maintaining cellular homeostasis (Dikic and Elazar, 2018; Bharath et al., 2021). Conversely, dysfunctional autophagy fails to maintain normal cellular protection and may, in turn, exert a dual effect, damaging cells and leading to various diseases such as heart disease, neurodegeneration, and metabolic disorders (Parzych and Klionsky, 2014). Hence, autophagy holds promise as a potential alternative for DMC therapy.
In recent decades, natural products (NPs) sourced from plants, animals, and microorganisms have attracted increased attention given their multifaceted components, ability to target multiple pathways, accessibility, and low toxicity levels (Newman and Cragg, 2016; Newman and Cragg, 2020). Recent studies have increasingly demonstrated the therapeutic potential of NPs in DR, DKD, DC, and DPN by modulating autophagy (Yao et al., 2018; Zhou P. et al., 2019; Yin et al., 2021; Liu P. et al., 2023; Liu T. et al., 2023). This article reviews recent research on autophagy and DMC, exploring the role and mechanism of NPs in enhancing DMC efficacy by regulating autophagy. Furthermore, NPs with autophagy-modulating abilities are highlighted as a promising therapeutic strategy for treating DMC.
2 AutophagyThe key to autophagy lies in the formation of autophagosomes and their subsequent fusion with lysosomes (Bharath et al., 2021). When the signal to regulate autophagy is received by the cell, various organelles within the cytoplasm generate an isolation membrane, also known as the phagophore. This membrane participates in the formation of phagosomes (Sanchez-Wandelmer et al., 2015; Morishita et al., 2017; Jung et al., 2020). The phagophore continues to elongate, encompassing the cell’s cytoplasm, damaged organelles, and long-lived proteins, eventually maturing into an autophagosome with a bimodal structure. Autophagosomes and lysosomes are transported by microtubules (MTs) to form autolysosomes (Korolchuk et al., 2011; Korolchuk and Rubinsztein, 2011). These autolysosomes degrade the inner membrane and contents, thereby providing for the cell’s needs and facilitating organelle renewal.
2.1 Classification of autophagyIn mammalian cells, autophagy can be classified into macrophage-, microautophagy-, and chaperone-mediated autophagy (CMA) according to different modes of entry into the lysosome (Habshi et al., 2023). Macrophages are characterized by the presence of a large autophagosome measuring about 500 nm. The phagosomal membrane under macrophagy extends, forming autophagosomes with a two-layer vesicular structure that encapsulates cytoplasmic components and fuses with lysosomes. Conversely, microautophagy directly segregates and internalizes cytoplasmic components by inwardly invaginating the lysosomal membrane (Schuck, 2020). Two types of microautophagy have been identified: 1) lysosomal membrane invagination (or endosomal membrane in the nucleus microautophagy) and 2) lysosomal membrane protrusion (Wang et al., 2023). While the former predominates in mammals, Komagataella phaffii illustrates the typical lysosomal membrane protrusion in microautophagy. CMA involves the specific recognition of substrate protein molecules containing the KFERQ group by the molecular chaperone protein Hsc70 (also known as HSPA8). These complexes then bind to the lysosomal membrane receptor LAMP2A, ultimately resulting in substrate degradation within lysosomes (Hubert et al., 2022). Macroautophagy involves membrane elongation, and microautophagy involves membrane invagination. Unlike macroautophagy and microautophagy, CMA does not entail membrane deformation (Kaushik and Cuervo, 2018) (Figure 1).
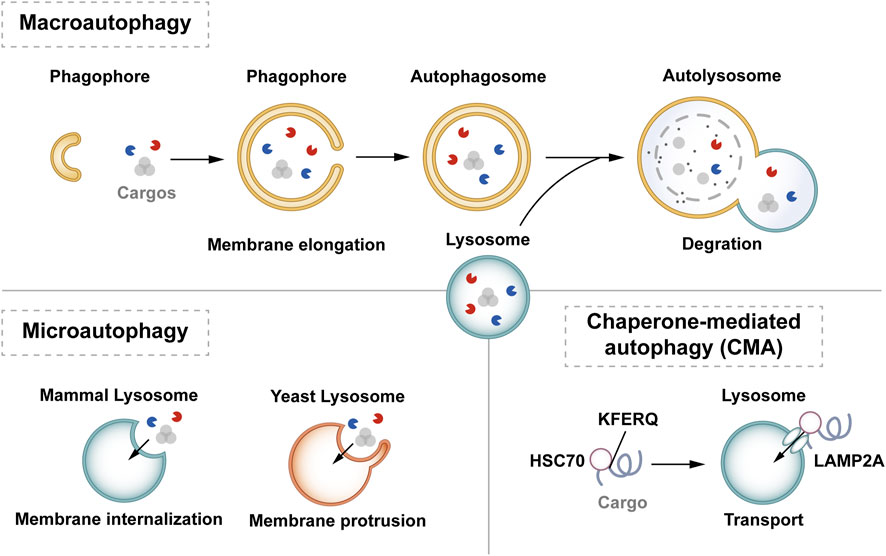
Figure 1. Classification of autophagy. Autophagy can be classified into three categories according to its cargos delivery pathway and membrane dynamics: macroautophagy, microautophagy, and CMA. Macroautophagy involves membrane elongation, microautophagy involves membrane invagination, and CMA does not entail membrane deformation. Hsc70 specifically recognises the KFERQ motif and then binds to the lysosomal membrane receptor LAMP2A to complete the autophagy process.
According to substrate selectivity, autophagy can be divided into non-selective autophagy and selective autophagy (Jing and Lim, 2012; Vargas et al., 2023). Non-selective autophagy mainly refers to autophagy that is induced in most cases, such as autophagy induced by mammalian target of rapamycin (mTOR) (Glick et al., 2010). In selective autophagy, specific autophagy adapter proteins like SQSTM1/p62, interact with microtubule-associated protein light chain 3 (Lc3) located in the phagolysosome membrane, delivering substrates directly to autophagosomes for degradation (Lee et al., 2023). Identified types of selective autophagy are mitophagy, ribosomal autophagy, endoplasmic reticulum (ER) autophagy, and peroxisome autophagy (Adriaenssens et al., 2022). Both macroautophagy and microautophagy can exhibit selective or non-selective behavior. Macroautophagy is considered the main type of autophagy and is the most widely studied type. Therefore, hereafter in this review, we refer to macroautophagy as “autophagy”.
2.2 The process of autophagyThe essence of autophagy is actually intracellular membrane rearrangement, which occurs as a dynamic process called autophagic flux and occurs in the following four broad stages: initiation of autophagy → formation of phagosomes and autophagosomes → fusion of autophagosomes with lysosomes → cleavage of autophagosomes (Figure 2).
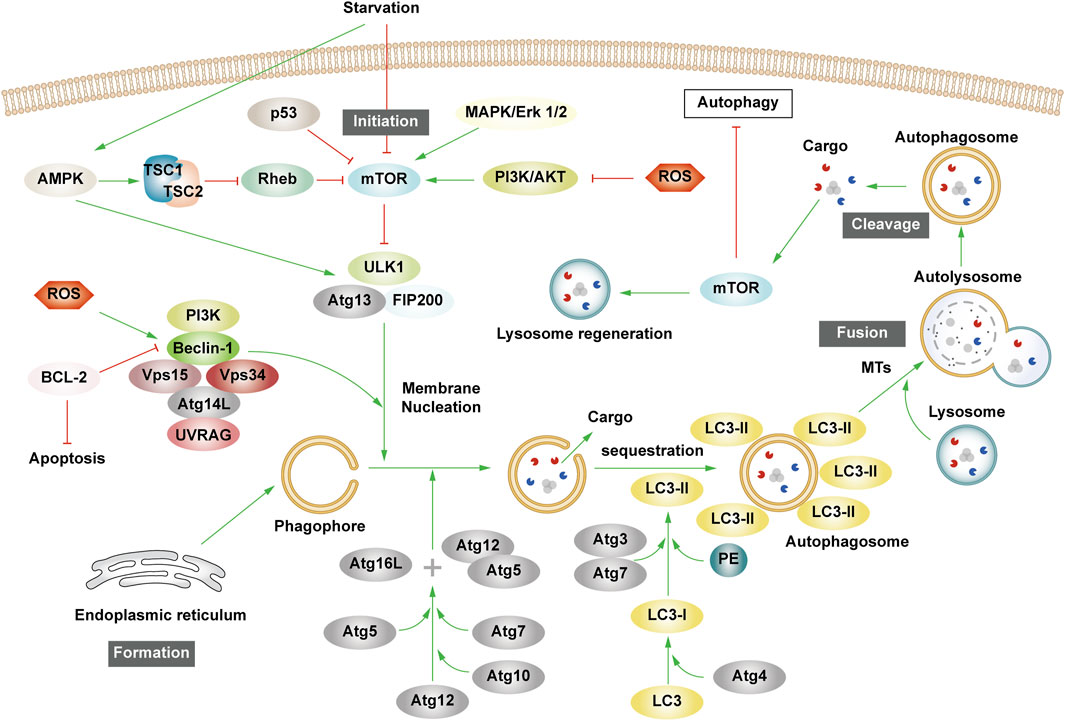
Figure 2. The process of autophagy. Green arrows indicate downstream cellular events; red lines indicate inhibition. Four major steps are involved in the process of autophagy: initiation, formation, fusion, and cleavage. Initiation: External stimuli such as starvation can affect the AMPK and mTOR pathways, leading to the activation of the ULK1 complex. Formation: The autophagosome formation is aided by two ubiquitin-like Atg-coupled systems and the PI3K complex. Fusion: Autophagosomes and lysosomes undergo transport via MTs to form autolysosomes. Cleavage: Degradation of cargos triggers autophagic lysosomal reorganization.
2.2.1 Autophagy initiationAutophagy is triggered in response to cell stress and can be considered as a coping mechanism to maintain homeostasis (Kume and Koya, 2015). The Ser/Thr signaling kinase mTOR regulates autophagosome formation. Pathways such as Akt and mitogen-activated protein kinase (MAPK) signaling pathways that activate mTOR inhibit autophagy, while those negatively regulating mTOR, such as the adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and p53-signaling pathways, promote autophagy (Alers et al., 2012). The ULK1 complex, comprising ULK1 or ULK2, FIP200, and mAtg13, forms a bridge in vivo between the upstream nutrient or energy receptors mTOR and AMPK and the downstream formation of autophagosomes (Leventhal et al., 2017). Under starvation conditions, AMPK is activated, mTOR is inactivated, and AMPK activation catalyzes ULK1 phosphorylation, thereby promoting autophagy. Conversely, in nutrient-rich conditions, AMPK is inactivated and mTOR binds to ULK1 serine 757 to inhibit the ULK1-AMPK interaction, leading to the inactivation of ULK1 and the eventual inhibition of autophagy (Alers et al., 2012). Interestingly, AMPK can inhibit Rheb, an mTORC1 activator, by activating Tuberous Sclerosis 1/2 (TSC1/2), which can then initiate autophagy (Inoki et al., 2006). Reactive oxygen species (ROS) can inhibit autophagy by suppressing TSC1/2 via the phosphatidylinositol-3-kinase (PI3K)/AKT pathway.
2.2.2 Phagosome and autophagosome formationUpon receiving autophagy regulation signals generated by cellular stress, phagosomes nucleate, expand, surround the cytoplasm, and finally form autophagosomes. Although the mechanism of phagosome formation is well understood, the constituent lipids and membrane modeling proteins involved in determining the shape and size of phagosomes have been at the center of controversy for decades (Tooze, 2013; Carlsson and Simonsen, 2015). It is now widely accepted that phagosomes originate near or on the ER, where several organelles including the mitochondria, Golgi complex, plasma membrane, and endosomes provide membranes for phagosome formation (Graef et al., 2013; Shibutani and Yoshimori, 2014; Sanchez-Wandelmer et al., 2015; Li J. et al., 2021). It has also been found that the ER exit site works synergy Atg9 in the autophagy-related gene (Atg) family to promote the assembly of autophagy mechanism, and may provide membranes for phagophore nucleation, maturation, and growth (Graef et al., 2013; Tooze, 2013; Sanchez-Wandelmer et al., 2015).
Phagosome nucleation hinges on local phosphatidylinositol 3-phosphate (PI3P) production. Regulated ULK complex recruitment to the phagocyte nucleation site phosphorylates beclin-1, activating the PI3K complex involving class III PI3K (PIK3C3), Beclin-1, VPS15, and Atg14L (Funderburk et al., 2010; Dooley et al., 2014).
Post-phagosome nucleation, membrane expansion involves two ubiquitin-like coupling systems: the Atg12 coupling system and Lc3-phosphatidylethanolamine (PE) coupling system (Pohl and Dikic, 2019). The Atg12 coupling system links Atg12 to Atg5 through Atg7 and Atg10 (E1-and E2-like enzymes), subsequently forming the Atg12-Atg5-Atg16 complex that contributes to LC3- and PE-coupling reactions. After the synthesis of LC3 protein, the C-terminal 5 peptide was cut-off by Atg4, resulting in cytoplasmic localization of LC3-I. LC3-I of LC3 was coupled with PE by ubiquitin-like reaction of Atg7 and Atg3 (E1-and E2-like enzymes, respectively) to form LC3-II. LC3-II stays on the inner and outer membrane of autophagosomes and serves as a marker of autophagy. Its upregulation is an indicator of autophagy activation and autophagosome formation (Glick et al., 2010). Post-autophagosome-lysosome fusion, LC3-II on the inner membrane is lysosomally degraded, and LC3-II on the outer membrane is cleaved by Atg4 to produce LC3-I for recycling (Mizushima and Komatsu, 2011).
2.2.3 Autolysosome production and cleavageMTs, as an internetwork of intracellular movement, are driven by specific kinesins, including the kinesin and cytoplasmic dynein (Kast and Dominguez, 2017). The interaction between autophagosomes dispersed in the cytoplasm and lysosomes enriched in the perikaryon depends on their two-way movement on microtubules. After maturation in the cytoplasm, autophagosomes are regulated by dynein to move to the perinuclear region. Under various stress conditions, intracellular pH rises, leading to the migration of lysosomes to the perinuclear region (Gross et al., 2007; Kimura et al., 2008). Autophagosomes and lysosomes in the same region fuse to form an autolysosome.
Upon autolysosome formation, lysosomal hydrolases degrade substrates, generating amino acids that reactivate mTOR via negative feedback, inhibit autophagy, regenerate the lysosome, and restore lysosomal levels—a process termed autophagic lysosome reorganization (Chen and Yu, 2018).
3 The molecular mechanism of autophagy and diabetic microvascular complications3.1 Interaction between autophagy and cellular biological processes related to diabetic microvascular complicationsPatients experiencing DMC endure chronic hyperglycemia, fostering abnormalities in nutrient perception, ER stress, oxidative stress, inflammation, and apoptosis. This persistent state leads to the accumulation of damaged proteins and organelles over time, jeopardizing cellular physiological functions. Autophagy, a scavenging mechanism, plays a pivotal role in maintaining cellular homeostasis and combating DMC by influencing these biological processes. (Figure 3).
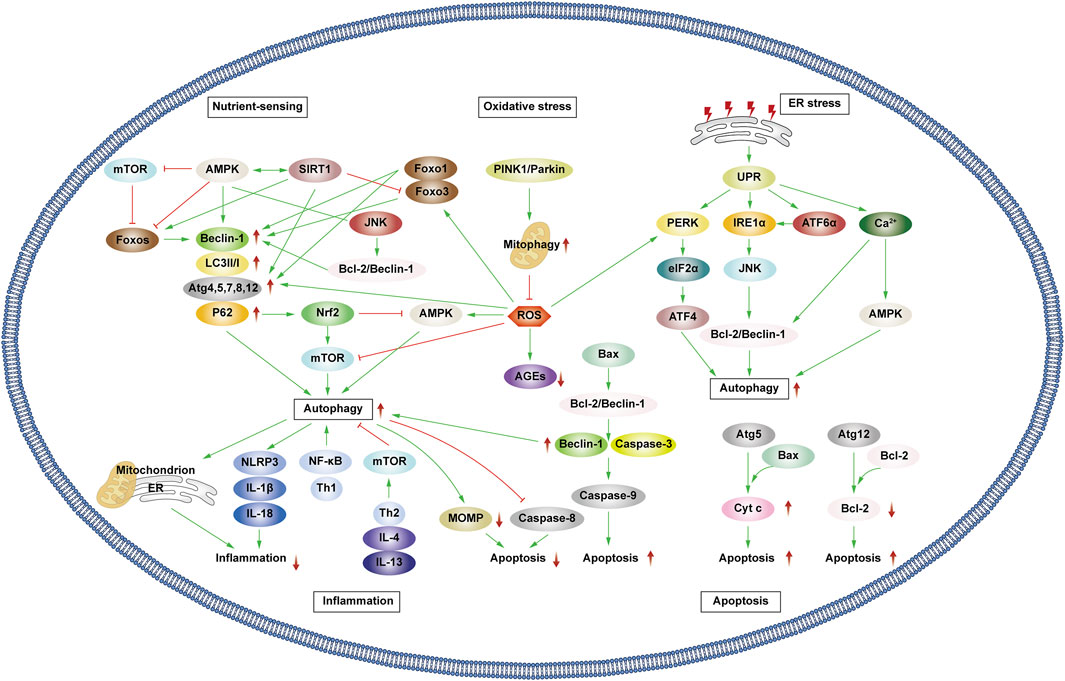
Figure 3. Autophagy and cellular biological processes. Green arrows indicate downstream cellular events; red lines indicate inhibition. Cellular biological processes mainly include nutrient sensing, oxidative stress, ER stress, inflammation and apoptosis. Cellular biological processes can regulate autophagy, which in turn can influence intracellular oxidative stress, inflammation, and apoptosis.
3.1.1 Interaction between autophagy and nutrient sensingHunger is known to promote autophagy through nutrient-sensing pathways, including mTOR, AMPK, and Silent Information Regulator 1 (SIRT1) (Oza et al., 2021). Briefly, as outlined earlier, mTOR and AMPK exhibit inhibitory and promotional effects on autophagy. SIRT1, a histone deacetylase, senses cellular energy levels and has the potential to slow cellular aging, withstand external stress, and enhance metabolism. SIRT1 activates autophagy via deacetylation of FOXOs. Additionally, it forms molecular complexes by directly binding to Atg5, Atg7, and Atg8, restoring autophagy (Ganesan et al., 2017; Gautam et al., 2020; Zhou et al., 2021). Moreover, SIRT1 deacetylates p53, reducing p53 expression, hence activating autophagy. The regulation of p53 in autophagy depends on its cellular localization, contributing to its dual role (Lee, 2019; Cui et al., 2021). Additionally, SIRT1 activates AMPK by deacetylating liver kinase B1 (LKB1). In turn, AMPK promotes the activation of SIRT1. Under starvation conditions, SIRT1 expression is elevated, activating autophagy in response to nutrient deprivation.
3.1.2 Interaction between autophagy and oxidative stressProlonged hyperglycemia results in oxidative stress within DMC-associated cells, leading to ROS accumulation and cellular oxidative damage. ROS accumulation triggers autophagy to reduce ROS levels, maintaining cellular homeostasis. ROS upregulate AMPK, FOXOs, MAPK, and c-JunN-terminal kinase (JNK) pathways or inhibit the PI3K/AKT/mTOR pathways, activating autophagy (Wong et al., 2010; Li et al., 2015; Hinchy et al., 2018; Kim et al., 2018). Notably, ROS influences Atg4 and ER stress pathways, promoting autophagy (Scherz-Shouval et al., 2007; Wible and Bratton, 2018). Primarily produced by damaged mitochondria, ROS levels are reduced by autophagy, particularly mitophagy, which selectively eliminates dysfunctional mitochondria, and help in maintaining cellular homeostasis. Additionally, p62 affects the Kelch-like ECH-associated protein 1 (Keap1)/Nuclear factor erythroid 2 related factor 2 (Nrf2) pathway, stimulating Nrf2 expression and enhancing mitophagy to counteract ROS (Wible and Bratton, 2018).
Mitophagy, chiefly regulated by the PTEN-induced kinase 1 (PINK1)/Parkin pathway in mammals, selectively removes damaged or redundant mitochondria. Upon mitochondrial depolarization, PINK1 rapidly recruits and activates Parkin to ubiquitinate the mitochondrial membrane. This ubiquitination, identified by p62, facilitates transportation to autophagosomes via LC3, ultimately leading to degradation (Lu et al., 2023). Beyond mitophagy, FOXOs associated with oxidative stress also impact autophagy.
The FOXO transcription factor family regulates various cellular physiological processes such as apoptosis, glucose metabolism, oxidative stress resistance, and lifespan extension (Ou et al., 2021). FOXO1 and FOXO3 are the most widely utilized members of the FOXO family, which can combine with promoter regions to activate autophagy genes such as Vps34, Beclin-1 and Rab7 (Ferdous et al., 2010; Webb and Brunet, 2014). Furthermore, they directly interact with autophagy proteins such as Atg7 and Atg4 (Cheng, 2019; Zhang X. et al., 2022). AMPK and PI3K/AKT promote FOXO phosphorylation, prompting their translocation from the nucleus to the cytoplasm, impeding normal transcriptional activity and thereby affecting autophagy (Puthanveetil et al., 2013; Zhou G. et al., 2019). Additionally, STRT1’s deacetylation of FOXOs triggers autophagy (Zhou et al., 2021).
3.1.3 Interaction between autophagy and ER stressAbnormal mitochondrial function and ER stress response are potential causes of insulin resistance (IR) in DMC patients. Autophagy, crucial in maintaining mitochondrial function and influencing ER stability, plays a pivotal role (Jung and Lee, 2010). The ER is the main part of protein folding as well as the main storage organelle of intracellular Ca2+. In response to the persistent hyperglycemic state and the corresponding cellular damage, cells in DMC patients, such as pancreatic β cells, produce large amounts of associated stress proteins, including insulin, which exceeds the ability of the ER to eliminate misfolded proteins, leading to the accumulation of a large number of unfolded or misfolded proteins that can trigger the unfolded protein response (UPR), thus resulting in ER stress and even DMC-related cell apoptosis (Yong et al., 2021). The UPR defends cells by degrading misfolded proteins, halting protein translation, and enhancing molecular chaperones for protein folding through endoplasmic reticulum-associated degradation (ERAD), and ER autophagy (inclusive of ER stress-mediated autophagy and ER-phagy) (Yong et al., 2021). UPR is mainly regulated by three ER-transmembrane proteins: PKR-like eukaryotic initiation factor 2α kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6α (ATF6α). PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α), curbing protein synthesis and reducing the amount of protein entering the ER. IIRE1α and ATF6α facilitate UPR target gene transcription such as foldase (Park et al., 2018). Autophagy regulation involves PERK’s phosphorylation of eIF2α, ATF4 induction, and increased Atg5 and Atg12 expression, thereby enhancing autophagy. IRE1α activates JNK, phosphorylates anti-apoptotic Bcl-2, ruptures Beclin-1/Bcl-2 complexes, and activates autophagy. ATF6α indirectly triggers autophagy by affecting IRE1α. Furthermore, Ca2+ activates the CaMKK/AMPK pathway during ER stress, promoting Beclin-1 dissociation from Bcl-2, thus enhancing autophagy. ER-phagy is a form of selective autophagy that relies on LC3 and Atg8 to eliminate damaged portions of the ER to sustain ER homeostasis (Senft and Ronai, 2015).
3.1.4 Interplay between autophagy and inflammationAutophagy interacts with inflammation. The cytoplasmic clearance of autophagy exhibits anti-inflammatory effects in any cell capable of activating autonomic inflammation. Moreover, the regulation of mitochondrial and ER function by autophagy affects the functioning of immune cells and influences the onset and resolution of inflammation (Deretic, 2021). Conversely, inflammation can activate or inhibit autophagy through different pathways, resulting in the suppression or exacerbation of inflammatory responses.
Inflammasomes are protein complexes activated by external or internal stimuli and can trigger inflammatory responses. The NLRP3 inflammasome is the most extensively researched inflammasome. In obese mice and humans, the NLRP3 inflammasome is upregulated, leading to the expression of TNF-α, a cytokine that is well-known to promote IR. Thus, NLRP3 inflammasome expression levels correspond with Type 2 diabetes mellitus (T2DM) severity (Komalla et al., 2020; Yang et al., 2020). The production of NLRP3 is closely related to the excessive accumulation of ROS, ER dysfunction, and the corresponding calcium outflow. Autophagy regulates these processes to inhibit the activation of inflammasomes and the pro-inflammatory cytokines IL-1β and IL-18. Specific NLRP3 components targeted by P62/SQSTM1 affect inflammatory mediator expression, thereby controlling inflammation. Studies have shown that NLRP3 production can interact with Beclin-1 to promote LC3-II expression and activate autophagy (Biasizzo and Kopitar-Jerala, 2020). Various inflammatory mediators, including NF-κB, Toll-like receptors, ROS, and Th1 cytokines enhance autophagy. Conversely, Th2 and anti-inflammatory cytokines like IL4 and IL13 inhibit autophagy by activating mTOR (Lapaquette et al., 2015).
In patients with DKD, the accumulation of advanced glycation end products (AGEs) and IL-1β accumulation incite inflammation, exacerbating disease progression. Transcription factor EB (TFEB) is a regulator of lysosomal biogenesis. Autophagy regulates the nuclear translocation of TFEB, enhancing lysosomal biogenesis and stimulating the degradation of AGEs in renal tubular cells (Kimura et al., 2017). In addition, numerous studies have shown that autophagy impairment in retinal cells also activates the inflammasome, leading to DR-related symptoms (Lin and Xu, 2019).
3.1.5 Interplay between autophagy and apoptosisType I cell death is the direct result of apoptosis, while type II programmed cell death occurs when autophagy overconsumes most of the organelles. Mitochondrial outer membrane permeabilization (MOMP) is a crucial step in the apoptosis process. Upon external stimulation, the level of anti-apoptotic protein Bcl-2 decreases, leading to the recruitment of pro-apoptotic protein Bax to the mitochondrial membrane that in turn activates MOMP. This results in the release of apoptotic proteins such as cytochrome c (Cyt c), apoptosis-inducing factor (AIF), and downstream-related apoptotic protein family caspase, which are necessary to complete the apoptosis process (Han et al., 2019). Autophagy can work alongside apoptosis to promote cell death. Additionally, autophagy is involved in some ATP-dependent apoptosis processes. However, there is also an antagonistic relationship between autophagy and apoptosis. Autophagy maintains mitochondrial homeostasis, scavenges ROS, reduces ER stress, and mitigates oxidative stress and inflammation to protect cells (D'Arcy, 2019).
Various stress stimuli can co-activate autophagy and apoptosis, and they share multiple regulatory molecules that can even coordinate their transformation. Bcl-2 binds to Beclin-1 to form complexes that inhibit autophagy while maintaining anti-apoptotic effects. However, when Bax competitively binds Bcl-2, Beclin-1 is released, leading to the activation of autophagy. Caspase-3 inactivates Beclin-1 to inhibit autophagy, while Beclin-1 promotes apoptosis by increasing caspase-9 activity. Following cleavage, Atg5 can bind to Bax, inducing the production of cytochrome c and promoting apoptosis. Atg12 can bind to Bcl-2, inhibiting its content and enhancing apoptosis. Additionally, autophagy can eliminate damaged mitochondria, preventing MOMP and the removal of caspase8, which also prevents apoptosis (Mariño et al., 2014).
Research has shown that apoptosis of damaged nerve cells such as Schwann cells is modified following peripheral nerve injury. After treatment, there was an observed upregulation of autophagy, inhibition of apoptosis, and reduction of nerve pain. Proinflammatory cytokines may promote apoptosis and induce nerve pain, while autophagy can inhibit proinflammatory cytokine activity to target these processes (Liao et al., 2022). Excessive apoptosis contributes to coronary atherosclerosis and myocardial ischemic injury. Regulation of autophagy protects cardiomyocytes. Thus, regulation of the relationship between autophagy and apoptosis may be a mechanism by which the progression of heart disease is slowed (Dong Y. et al., 2019).
3.2 Autophagy and diabetic kidney diseaseDKD is the renal impairment caused by prolonged hyperglycemia which can affect the entire kidney. It is one of the most perilous complications of diabetes. DKD is characterized by impaired glomerular filtration rate (GFR), proteinuria, and structural abnormalities like glomerular basement membrane thickening and mesangial dilatation. The intricate pathogenesis involves apoptosis, inflammation, oxidative stress, ER stress, and nutrient-sensing pathways (Al Mamun et al., 2021). The treatment options for DKD are presently limited (Dorotea et al., 2022). Considerable evidence has shown that autophagy plays a pivotal role in blood sugar stability and renal protection. Targeted autophagy offers potential DKD treatment avenues (Liu et al., 2019; Erekat, 2022).
3.2.1 PodocytesPodocytes, namely glomerular epithelial cells, play a key role in maintaining glomerular filtration barrier and preventing proteinuria. As terminally differentiated cells, podocytes generally do not replicate, and there are no new cells to repair and replace after injury, which leads to glomerular damage. Hence, autophagy is essential for the maintenance of podocyte homeostasis (Liu et al., 2019). Early in diabetes, autophagy protects podocytes by removing damaged components. Various stress reactions affect autophagy, leading to podocyte damage and DKD development. Experiments have observed changes in autophagy in diabetic foot cells, which provide a basis for this conclusion (Gonzalez et al., 2021).
Research by Jin et al. identified microRNA-486 (miR-486) in adipose-derived stem cell exosomes, inhibiting mTOR via Smad1 downregulation, elevating autophagy, and reducing podocyte apoptosis (Jin et al., 2019). Xiao et al. discovered that after administering the mTOR inhibitor rpapamycin to streptozotocin (STZ)-induced DM mice, there was a significant increase in LC3 expression in podocytes, upregulation of autophagy activity, and reduction in kidney injury (Xiao et al., 2014). Metformin, an AMPK agonist, restores autophagic activity in podocytes, protecting against DM injury (Song A. et al., 2021). Similarly, according to Xu et al., SIRT1/FOXO1 pathways influenced by metformin enhance autophagy, mitigating diabetic kidney injury (Xu J. et al., 2020). Su et al. found that the levels of podocyte autophagy-associated factors (Beclin-1, LC3), as well as the LC3B II/I ratio, decreased with time after high glucose (HG) stimulation and were particularly significant after 24 h. Overexpressed LncRNA AK044604 (regulator of insulin sensitivity and autophagy [Risa]) can reduce the levels of downstream autophagy-related proteins (Beclin-1 and LC3B) through the SIRT1/glycogen synthase kinase-3β (GSK-3β) axis in the HG environment, leading to podocyte damage, thereby accelerating the development of DKD (Su PP. et al., 2022).
3.2.2 Renal tubular epithelial cellsThe above-described experimental results confirm that podocyte autophagy will be damaged under long-term HG environment, which ultimately leads to DKD. In addition to podocytes, renal tubular epithelial cells (RTECs) play an important role in maintaining renal function. RTECs are the key part of renal reabsorption activity, and their active transport function consumes a large amount of energy, which makes them more susceptible to hypoxia or energy deprivation. Therefore, maintaining a normal level of autophagy ensures the survival of RTECs in nutrient-poor environments. Experimentally, Atg7 knockout mice demonstrated exacerbated DKD progression, highlighting autophagy’s protective role in RTECs. miR-214 suppression of ULK1 via proximal renal tubule knockout promotes autophagy, reducing renal hypertrophy and albuminuria (Ma et al., 2020). In Liu L. et al., 2022 research, RTECs were one of the most abundant cell types in the mitochondria, so it is more susceptible to mitochondrial disorder. PHB2 is a kind of mitophagy receptor. In STZ-induced DKD mice, PHB2 degradation via TNF-α promoted DKD progression by impairing mitophagy. Kitada et al., 2016 observed that providing obese Wistar rats a very low protein diet could inhibit the mTOR activity of RTECs, reduce renal inflammation, and slows the progression of advanced DKD.
3.3 Autophagy and diabetic retinopathyDR, one of the most common microvascular complications of diabetes, encompasses neurovascular changes leading to irreversible blindness. Today, DR is not just considered a simple microvascular disease, rather one that involves neurodegenerative changes. The American Diabetes Association redefines DR as a neurovascular complication of diabetes. The progression involves pericyte loss, microangiomas, capillary non-perfusion zones, vascular endothelial growth factor (VEGF) secretion, and fragile capillary formation, thereby contributing to vitreous hemorrhage and retinal detachment (Yang et al., 2022). Under the induction of long-term HG level, oxidative stress overload, inflammation, ER stress, and autophagy disorder in the retina lead to retinal microvascular damage and glial cell degeneration, including blood-retinal barrier (BRB) changes, retinal neuron dysfunction and neuronal apoptosis, finally developing into DR (Adornetto et al., 2021).
Autophagy can protect the retina by eliminating ROS, reducing ER stress, apoptosis, and the production of proinflammatory factors (Russo et al., 2013). In fact, neuron cells, podocytes, and RTECs share high similarities. Neurons and podocytes, as highly differentiated cells, cannot undergo the next step of mitosis. At the same time, neurons are metabolically as active as RTECs. Therefore, autophagy disorder is also closely related to neurotoxicity and neurodegeneration. Autophagic activity under mild stress plays a protective role for cell survival, whereas dysregulation of autophagic activity leads to massive cell death, giving rise to the deterioration of DR (Gong et al., 2021).
3.3.1 Blood-retinal barrierA key feature of DR is the decomposition of the BRB, which is composed of the retinal vascular system (inner layer) and retinal pigment epithelium (RPE) cells (outer layer). The former comprises retinal capillary endothelial cells, peripheral cells, and the basement membrane. The latter is conductive to maintaining retinal fluid balance and photoreceptor function (Gong et al., 2021). After human retinal pigment epithelial cells (ARPE-19) were exposed to HG, treatment with arbutin (a naturally occurring soluble glycosylated phenol) increased SIRT1 expression, enhanced cellular autophagy, and inhibited inflammation and apoptosis (Ma et al., 2021). However, some researchers have found that the inhibition of autophagy can delay DR. Fen et al. found that impairment of autophagic degradation is usually accompanied by the accumulation of p62. The level of p62 in the retina of STZ-induced diabetic rats was significantly higher than that of non-diabetic rats, suggesting errors in autophagic degradation. They suggested that this was caused by the induction of lysosomal membrane permeability (LMP) in HG, resulting in the inability of lysosomes to function properly. Under HG conditions, the downregulation of high mobility group box 1 (HMGB1) rescued LMP, increased mitochondrial electrochemical potential (ΔΨm), restored autophagic degradation, reduced ROS production and VEGF and inflammatory factor expression, and ultimately protected RPE cells (Feng et al., 2022). In another experiment, exposure to HG conditions decreased ARPE-19 cells viability, increased apoptosis and LC3-II/LC3-I, p-p53 protein expression, decreased p62 and p-mTOR expression, and increased autophagic flux. Treatment with proanthocyanidins, a polyphenol compound, weakened the above changes. When rapamycin was added, apoptosis was again increased in the proanthocyanidins-treated ARPE-19 cells, suggesting that increased autophagy in the HG environment impairs ARPE-19 cells and that proanthocyanidins reverse this injury (Li et al., 2022).
3.3.2 Retinal ganglion cellsRetinal ganglion cells (RGCs), the only afferent neurons responsible for transmitting visual information to the visual center, are one of the main cells that maintain visual function. In DR, the levels of p-mTOR and its downstream Ps6 in RGCs were upregulated in STZ-induced mice in the first month, and the cells were slightly lysed. Then, p-mTOR and Ps6 showed a downward trend after 2 months, and their expression reached the lowest level after 6 months, while the number of apoptotic cells peaked. This trend could be changed by adding rapamycin, indicating that mTOR inactivation is an important factor in RGCs damage and that RGCs damage aggravates the course of DR (Madrakhimov et al., 2021). Another study also proved that autophagy damage induced by HG led to RGCs apoptosis and aggravated DR. Compared with the normal group of rat retinal precursor R28 cells, the late apoptosis level in the HG group was significantly higher, and the early apoptosis level in the HG group was also higher than that in normal group (Peng et al., 2022). Most glial cells are Müller cells that exist in all retinal layers; they can provide energy for neurons and are also the main source of VEGF. Wang Y. et al., 2020 showed that the expression of Beclin-1, Atg7, and LC3-IIof Müller cells in the retina of STZ-induced diabetic mice were lower than those in the control group. Autophagy could be promoted by inhibiting the mTOR pathway, which could inhibit the secretion of inflammatory factors and retard the progression of DR. Other studies show that the expression of Beclin-1 and LC3-II were upregulated in Müller cells under HG, which indicated the existence of autophagosomes. However, the subsequent increase of ER stress and the accumulation of p62 indicated that lysosomal function was imbalanced under stress, and the cargos could not be degraded, leading to the release of VEGF and the apoptosis of Müller cells. After 3-methyladenine (3-MA) was used to inhibit autophagy, the number of apoptosed Müller cells increased. On the contrary, after rapamycin was used to restore autophagy degradation and prevent VEGF release from increasing to improve lysosomal proteolytic activity, Beclin-1 was upregulated and the number of apoptosed Müller cells decreased significantly (Lopes de Faria et al., 2016). According to Fu et al., the activation of AMPK after HOG-LDL stimulation in Müller cells led to a significant increase in the levels of autophagy-related proteins Atg5, Beclin-1, and LC3II/LC3I. At the same time, the expression of caspase-3 was further increased, indicating that the activation of autophagy would lead to apoptosis (Fu et al., 2016). In addition, studies have also shown that activation of AMPK can reduce the expression of apoptosis-related proteins Bax, increase Bcl-2, and decrease LC3 expression, and restore autophagy and mitochondrial function to delay DR-induced photoreceptor cell degeneration (Song S. et al., 2021).
3.4 Autophagy and diabetic cardiomyopathyProlonged diabetes exacerbates diastolic and systolic heart failure post-myocardial infarction, resulting in myocardium-specific microvascular complications, significantly elevating mortality rates among diabetic individuals. DCM, with mild symptoms in the early stage, is difficult to treat in the later stage. Thus, comprehending DCM’s pathogenesis holds significant importance for early diagnosis. Accumulation of ROS is a pivotal pathogenic factor in DCM development, impeding myocardial contractility, and normal cardiac function through redox injury, mitochondrial dysfunction, apoptosis, and myocardial cell fibrosis (Tan et al., 2020). ROS accumulation also prompts aberrant autophagy, crucial for cardiac homeostasis across various cardiovascular cells. Studies indicate abnormal cellular metabolism and accumulation of damaged organelles in DCM rat cardiomyocytes. Similarly, preclinical trials involving DCM patients have demonstrated dysregulated autophagy (Kobayashi and Liang, 2015; Jia et al., 2018). Consequently, autophagy assumes a critical role in DCM. However, the precise influence of autophagy on DCM remains poorly understood. Various researchers have delineated both protective and pathogenic roles of autophagy in type 1 and type 2 diabetes-induced cardiomyopathy, respectively. Autophagy’s role may hinge on diabetes type, stage, and severity (Lee et al., 2018; Dewanjee et al., 2021). Some studies have proposed an “optimal interval” for autophagy in DCM, where optimal therapeutic effects are achieved (Xie et al., 2011; Russo et al., 2012).
3.4.1 Type 1 diabetic heartIn type 1 diabetes mellitus, insulin deficiency hampers signal transduction-regulated glucose and fatty acid transport, leading to cellular starvation, reduced ATP levels, and subsequent AMPK activation initiating autophagy. Mice studies that used chloroquine (CQ) to inhibit autophagy showed cardiomyocyte damage (Kanamori et al., 2015). However, contradictory findings also exist. Xu et al. propose autophagy inhibition as an adaptive response limiting heart damage in type 1 diabetes (Xu et al., 2013). Similarly, Yuan et al. reported alleviated myocardial apoptosis and cardiac fibrosis following CQ-induced autophagy inhibition in mice (Yuan et al., 2016). Thus, further experiments are warranted to elucidate the impact of autophagy in a type 1 diabetic heart.
3.4.2 Type 2 diabetic heartIn contrast to type 1 diabetes, studies suggest that inhibiting myocardial cell autophagy in T2DM can enhance myocardial cell survival. Kanamori et al. found that although the expression of LC3-II was upregulated in db/db mice with T2DM, degenerated mitochondria and autophagosomes were observed in the heart of mice, and neither mature autolysosomes and nor a small amount of lysosomes were detected, which indicated the delay of autophagy flux. Thus, the increase of LC3-II accumulation was considered related to autophagy damage in the last digestive step (Kanamori et al., 2015). Additional research supports these findings. Kobayashi et al. observed reduced autophagic flux in cardiomyocytes cultured in HG, with decreased LC3-II expression. Inhibition of autophagy with 3-MA reduced cardiomyocyte mortality, while upregulating Beclin-1 or Atg7 rendered cardiomyocytes susceptible to HG toxicity (Kobayashi et al., 2012).
Some studies highlight the cardioprotective effects of enhancing autophagy in T2DM. Metformin’s ability to activate AMPK enhances autophagy, reducing myocardial fibrosis and hypertrophy while partially reversing left ventricular dilatation (Kanamori et al., 2019). Zinc supplementation in diabetic rat cardiomyocytes led to reduced LC3-II levels and restored cardiac function, indicating a potential protective role of zinc by inhibiting autophagy (Lu et al., 2015). Chen et al. demonstrated that AMPK pathway activation via the helix B surface peptide (HBSP) in STZ-induced mice increased the LC3-II/LC3-I ratio, reduced p62 levels, inhibited myocardial cell apoptosis, and restored cardiac function. Conversely, using 3-MA diminished the HBSP-induced benefits (Lin et al., 2017).
This conflicting perspective on autophagy-induced DCM represents the previously mentioned “optimal interval.” While autophagy activation is often deemed cardioprotective, excessive autophagy can culminate in cell death. Hence, careful timing and dosage consideration of autophagy as a therapeutic target for DCM are pivotal for optimal therapeutic effects.
3.5 Autophagy and diabetic peripheral neuropathyDPN is a chronic diabetes-related complication characterized by symmetrical distal limb numbness, tingling, muscle weakness, and nociceptive hypersensitivity. As the disease progresses, it can lead to severe consequences such as diabetic foot ulcers, gangrene, and amputation. The neural tissue in patients with DPN undergoes various alterations including hyperglycemia, ROS-induced oxidative stress and inflammation. These changes lead to dysfunction of the mitochondria and ER, which exacerbates the accumulation of harmful substances in neuronal cells and can even lead to apoptosis (Ardeleanu et al., 2020). Neuronal cells, being highly sensitive, are deeply affected by autophagy. Thus, autophagic clearance of harmful neuronal substances remains a key pathway for maintaining neuronal homeostasis.
Schwann cells (SCs) are nerve fibers in the peripheral nervous system that rely heavily on autophagy under normal physiological conditions to eliminate damaged myelinated cell fragments, aiding in nerve regeneration and repair. In DPN, demyelination and SC dysfunction resulting from exposure to an HG environment is noted, which induces critical pathological changes (Wang L. et al., 2020). HG-induced DPN mice and SCs showed elevated ROS levels, reduced LC3-II/LC3-I ratio, elevated p62 expression, and altered myelin thickness; moreover, axon contraction in the sciatic nerve indicated ROS-induced autophagy dysfunction in SCs as a potential cause of DPN (Choi et al., 2023). Yang et al. corroborated decreased autophagosome formation and downregulated sciatic nerve Beclin-1 in STZ-induced rats, correlating with axon end expansion and Purkinje cell degeneration. Adding an autophagy inducer increased Beclin-1 expression in the sciatic nerve, enhancing SCs autophagy, ameliorating myelin degeneration, and alleviating axon atrophy (Yang et al., 2014). Moreover, Yuan et al. revealed decreased Beclin-1 and Atg3 expression in HG-stimulated SCs, yet stilbene lycorine via the AMPK pathway boosted autophagy, slowing DPN progression (Yuan et al., 2022). Additionally, translocator protein (TSPO) agonists displayed therapeutic effects on DPN, although their exact mechanism remains unclear. STZ-induced rats in the control group exhibited reduced Beclin-1 and LC3-II/LC3-I ratios in SCs. TSPO treatment increased Beclin-1 and LC3-II/LC3-I ratios while reducing p62 accumulation. However, these beneficial effects were nullified when using 3-MA, suggesting TSPO’s therapeutic role through autophagy modulation in SCs (Gao et al., 2023).
Some studies highlight that excessive autophagy can induce apoptosis and neuronal damage. In the STZ-induced DPN rat model, the expression of Lipin1 (a phosphatidic acid phosphatase, which is involved in maintaining normal peripheral nerve conduction function) was downregulated, and excessive autophagy was observed, which may lead to the increase of SCs apoptosis and the demyelination of sciatic nerves in DPN rats. Conversely, Lipin1 overexpression curtailed hyperglycemic-induced autophagy hyperactivity and apoptosis, ameliorating sciatic nerve pathology and motor nerve conduction velocity, thereby alleviating DPN (Wang et al., 2021). It is evident that while moderate autophagy exerts a positive influence on nerve repair and regeneration, excessive autophagy results in adverse effects.
4 Natural products for regulating diabetic microvascular complicationsOwing to the continued explorations of the mechanism of autophagy, its relationship with DMC is progressively becoming clearer. Despite claims of inadequate drug efficacy, investigating NPs with autophagy-regulating abilities offers new avenues for treating DMC. In recent years, NPs like flavonoids (e.g., apigenin, puerarin); polyphenols (e.g., paeonol, resveratrol); alkaloids (e.g., berberine [BBR]); disaccharide (e.g., trehalose); and amines (e.g., melatonin) have exhibited therapeutic potential in diseases via autophagy regulation. However, recognizing autophagy’s bidirectional regulatory effect on diseases mandates stringent evaluation and prediction of autophagy mechanisms, NP dosage, and action timing to ensure positive and stable therapeutic outcomes. This section elaborates on how NPs regulate autophagy through the mTOR, AMPK, and SIRT1 pathways. Additionally, it delves into NPs-mediated autophagy regulation in the mitochondria and ER, along with their roles in reducing oxidative stress, inflammation, and apoptosis.
4.1 Natural products and DKDThis section examines the use of NPs to target autophagy as a means of combating DKD. It covers various aspects of autophagy, including podocyte autophagy, renal epithelial cell autophagy, mitophagy, and other autophagic pathways. Additionally, it explores the interplay between autophagy, inflammation, and apoptosis (Figure 4; Table 1).
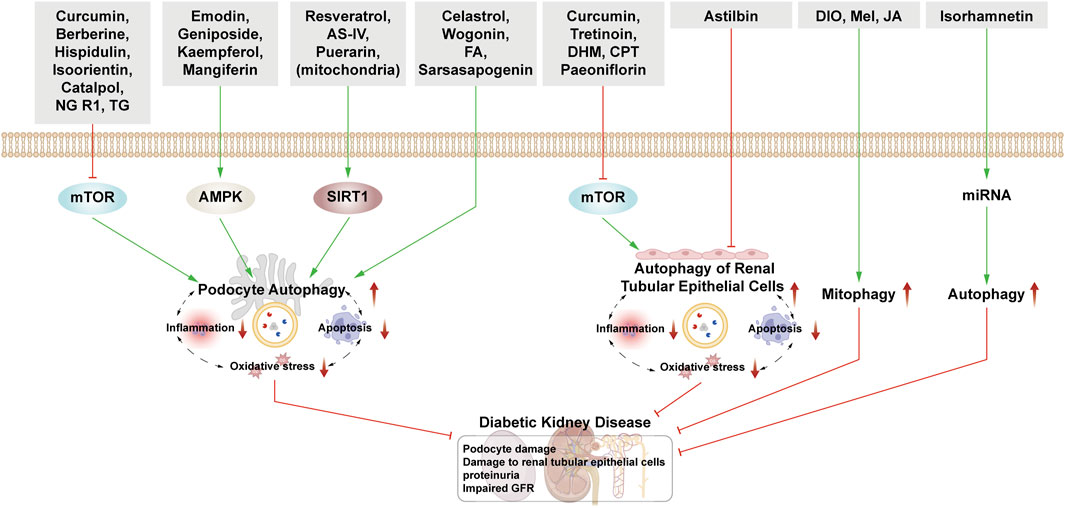
Figure 4. NPs target autophagy to fight DKD. Green arrows indicate downstream cellular events; red lines indicate inhibition. Related NPs target different nutrient-sensing and other pathways to activate autophagy in podocytes and renal epithelial cells, reducing oxidative stress, inflammation, and apoptosis. This helps alleviate the symptoms of DKD.
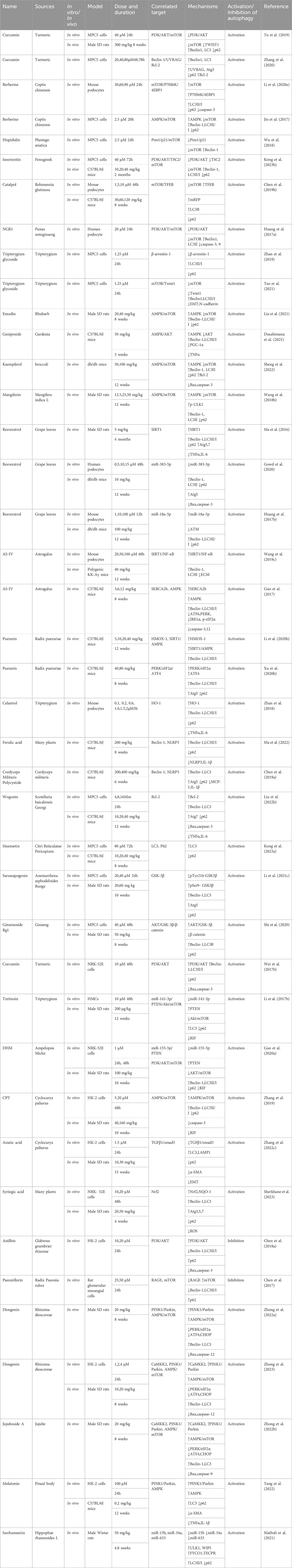
Table 1. NPs to target autophagy as a means of combating DKD.
4.1.1 Natural products mediate podocyte autophagy to alleviate renal injury4.1.1.1 Podocyte autophagy regulated by the mTOR pathwayCurcumin is derived from Curcuma longa and exhibits anti-inflammatory and anti-cancer properties. Podocyte epithelial mesenchymal transition (EMT) causes podocytes to fall off and be eliminated from the body. When EMT occurs, it is accompanied by the decrease of epithelial marker E-cadherin. In DKD rats, curcumin treatment significantly reduces blood glucose, urinary protein, and urea nitrogen levels. On a molecular level, it increases E-cadherin and LC3 protein expression while decreasing PI3K, p-Akt, and TWIST1. The expression of these proteins was reversed by the application of mTOR inducers. Therefore, it is suggested that curcumin can induce autophagy and reduce the changes of EMT in podocytes through the PI3k/Akt/mTOR pathway to treat DKD (Tu et al., 2019). Furthermore, Zhang et al., 2020 proved that after curcumin treatment, the expression of LC3, Beclin-1, UVRAG, ATG5, and Bcl-2 increased, while the expression of Bax and caspase-3 decreased, indicating that curcumin promoted podocyte autophagy and inhibited podocyte apoptosis by regulating Beclin-1/UVRAG/Bcl-2.
Berberine, a quaternary alkaloid isolated from Berberis vulgaris, is widely distributed in the plant kingdom and has antibacterial, anti-inflammatory, and antitumor properties. In HG-cultured mouse podocytes clone 5 (MPC5) cells, BBR activates autophagy by inhibiting the mTOR/P70S6K/4EBP1 signaling pathway, thereby safeguarding podocytes (Li C. et al., 2020). Moreover, BBR’s protective effect on podocytes involves increased Beclin-1 expression, LC3II/LC3I ratio, and autophagosome count via AMPK activation and mTOR inhibition. This effect is attenuated after 3-MA treatment, affirming BBR’s protective action on podocytes through autophagy (Jin et al., 2017).
Hispidulin is a natural flavonoid mainly extracted from Artemisia pilosula. Hispidulin possesses anti-inflammatory, hypoglycemic, and anti-angiogenesis properties. It induces autophagy by activating the Pim1/p21/mTOR signal axis, thereby mitigating HG-induced podocyte damage (Wu et al., 2018). Isoorientin, a flavonoid derived from the leaves of Bamboo and Charcot, has anti-oxidant and anti-inflammatory properties and improves IR. Under HG conditions, isoorientin stimulates autophagy via the PI3K/AKT/TSC2/mTOR pathway, significantly improving the damaged mitochondria’s autophagic clearance rate to protect podocytes. (Kong et al., 2023b).
Catalpol is a natural iridoid glycoside compound derived from Chinese medicinal herb Rehmannia glutinosa. It relieves renal pathological damage in DKD mice and rescues foot cytoskeletal destruction induced by HG. Catalpol enhances autophagy by inhibiting mTOR activity and promoting TFEB nuclear translocation, thus stabilizing podocyte cytoskeleton (Chen Y. et al., 2019). Notoginsenoside R1 (NGR1), derived from the dried roots and rhizomes of Panax notoginseng, is also a terpenoid. NGR1 protects podocytes from HG-induced injury by increasing autophagy, inhibiting apoptosis, and promoting cytoskeletal restoration via activation of the PI3K/Akt/mTOR pathway (Huang G. et al., 2017).
Tripterygium glycoside (TG), one of the active substances extracted from Tripterygium, has immunosuppressive and anti-inflammatory properties and has been used to reduce proteinuria in DKD patients. Zhan et al. showed that the effect of TG on podocytes is via both the activation of autophagy and the downregulation of β-arrestin-1 (Zhan et al., 2019). Mei et al. reported that overexpression of Twist1 could aggravate podocyte damage, and the podocyte damage was relieved after TG treatment. After adding mTORC1 activator, its therapeutic effect on podocytes was reversed. Therefore, TG can also activate autophagy through mTOR/Twist1 signaling pathway to alleviate EMT and podocyte apoptosis (Tao et al., 2021).
4.1.1.2 Podocyte autophagy regulated by AMPK pathwayEmodin is extracted from Pinelliae Rhizoma and Palmariae Rhizoma, and it reduces proteinuria and mitigates podocyte foot process fusion in DKD rats. Treatment with emodin increases LC3-II/I, p-AMPK, and Beclin-1 while reducing p62 and p-mTOR expression, suggesting rhodopsin-induced podocyte protection through enhanced autophagy via the AMPK/mTOR pathway (Liu et al., 2021).
Geniposide extracted from Gardenia is used to treat cardiovascular and cerebrovascular diseases as well as diabetes. In DKD mice, geniposide reduces podocyte loss, improves renal function, increases AMPK activity, and enhances autophagy. Moreover, it mitigates oxidative stress and inflammation in the kidney by reducing AKT activity (Dusabimana et al., 2021).
Kaempferol is a flavonoid and is widely present in fruits and vegetables. It alleviates mesangial matrix expansion and podocyte loss or fusion in DKD. It slows DKD progression by upregulating podocyte autophagy via the AMPK/mTOR pathway, evidenced by increased LC3II, Beclin-1, p-AMPK, Bcl-2, Atg7, and Atg5, and decreased p-mTOR, Bax, caspase-3, and p62 expression (Sheng et al., 2022). Mangiferin and phenolics from Physalis peruviana fruits have both been shown to exert a protective effect on the kidney by enhancing autophagy through the AMPK/mTOR pathway (Wang X. et al., 2018; Ezzat et al., 2021).
4.1.1.3 Podocyte autophagy regulated by SIRT1 pathwayResveratrol (RSV), a non-flavonoid polyphenol, can be synthesized in the leaves and skins of grapes and has antioxidant and anticancer properties and offers cardiovascular protection. RSV, as an activator of SIRT1 and AMPK, promotes the expression of SIRT1 under hypoxic conditions, up-regulating the expression of LC3, Atg7, and Atg5, and activates hypoxia-induced autophagy (Ma et al., 2016). Interestingly, RSV can inhibit ER stress by reducing the production of ROS and AGEs and activate AMPK to promote autophagy (Gowd et al., 2020). According to Huang et al. and Xu et al., RSV can inhibit miR-383-5p and upregulate miR-18a-5p, resulting in an increase in the ratio of LC3II to LC3I and a decrease in the expression of caspase-3 in podocytes, to inhibit foot cell apoptosis. The above protective effect of RSV can be inhibited by 3-MA, which further proves that RSV exerts therapeutic effect in DKD through autophagy (Huang SS. et al., 2017; Xu et al., 2017).
Astragaloside IV (AS-IV), extracted from Astragalus membranaceus, has good anti-virus and hypoglycemic effects. After intervention with astragaloside IV, the expression of SIRT1 is increased and the acetylation of NF-κB subunit p65 is decreased, reversing the EMT of podocyte, enhancing autophagy and protecting podocyte (Wang X. et al., 2019). Guo et al. also found that astragaloside IV improved the expression of sarcoendoplasmic reticulum Ca2+ ATPase 2b (SERCA2b) and activated AMPK, improving ER stress, activating autophagy and preventing the progress of DKD (Guo et al., 2017).
Puerarin extracted from radix puerariae, a leguminous plant, is often used in patients with coronary heart disease and hypertension. On the one hand, puerarin upregulates autophagy through HMOX1 and SIRT1, protecting podocytes and delaying the progression of DKD (Li X. et al., 2020). On the other hand, under conditions of ER stress, puerarin upregulates the PERK/eIF2α/ATF4 signaling pathway; increases the expression of Beclin-1, LC3II, and Atg5; downregulate
留言 (0)