
記住我

Sickle cell disease (SCD) affects over 100,000 individuals in the United States, involving a point mutation in the beta globin gene resulting in the production of sickle hemoglobin.1 This mutation can lead to painful vaso-occlusive crises or more life-threatening complications, such as acute chest syndrome, stroke, or myocardial infarction.2 Hepatic involvement in SCD is a rare and potentially fatal complication, seen primarily among those with homozygous sickle cell mutations.1,3,4 Cases of hepatic involvement in SCD have been sparsely documented in the literature, mostly described in children and with limited follow-up.1,5
The pathophysiology of sickle cell intrahepatic cholestasis (SCIC) involves sickling of sinusoidal hemoglobin within the liver, leading to intracanalicular obstruction, extensive hepatocyte necrosis, and dysfunction driven by cell hypoxia.1,2,6 Histologically, classic patterns include sickled red blood cells, ballooning hepatocytes, and dilated canaliculi with bile plugs.1
Clinically, SCIC often presents initially with abdominal pain, leukocytosis, and hyperbilirubinemia.1,2,6–8 SCIC can rapidly progress to renal and hepatic failure, the latter of which involves encephalopathy, coagulopathy, and marked transaminase elevation >1,000 mg/dL1,2,4,6,9,10
We present a case of acute liver failure secondary to SCIC, with the aim of highlighting the importance of early recognition and comparing various management strategies.
CASE REPORTA 32-year-old man with a history of sickle cell anemia, previously complicated by femoral head avascular necrosis and acute chest syndrome, presented to the hospital with chest pain similar to prior episodes of sickle cell crisis. The decrease in outside temperature was thought to be the trigger of symptoms. He was subsequently admitted for refractory symptoms and further pain control.
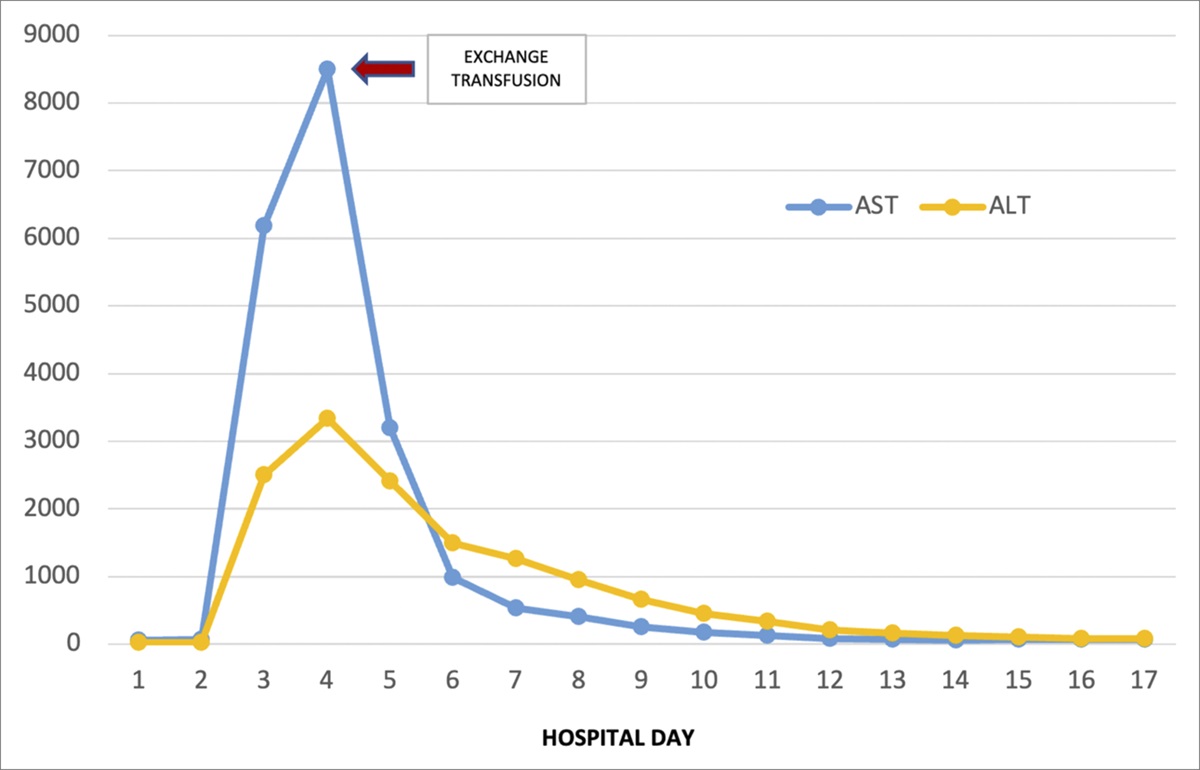
On admission, laboratory test results were as follows: aspartate transaminase (AST) 62, alanine transaminase (ALT) 21, Creatine (Cr) 0.63, white blood cells 11.6, hemoglobin 10.6, and platelets 301 (Table 1). By hospital day 3, his clinical status worsened, with new shortness of breath, abdominal pain, mild drowsiness evolving into encephalopathy, and an oxygen requirement of 5 L nasal cannula. Chest x-ray imaging demonstrated new opacities suggestive of acute chest syndrome. Simultaneously, the patient developed marked transaminase elevation (AST 6,188 and ALT 2,501), acute kidney injury (Cr 4.04), anemia (hemoglobin 7.6), leukocytosis (16.8), and elevated international normalised ratio (INR) (2.2). Abdominal ultrasound demonstrated cholelithiasis, hepatomegaly with coarsened echotexture, patent portal vein, and bilateral renal disease.
Table 1. - Trends in specific laboratory values in a patient with sickle cell intrahepatic cholestasis AST/ALT Cr WBC Total bilirubin Hemoglobin Day 1 62/21 0.63 11.6 7.0 10.6 Day 2 70/21 0.83 11.0 7.5 8.6 Day 3 6,188/2,501 4.04 16.8 22.6 7.6 Day 4 8,503/3,340 5.74 9.2 20.5 9.4 Day 5 3,190/2,410 6.75 7.9 28.4 11.4ALT, alanine transaminase; AST, aspartate transaminase; Cr, creatine; WBC, white blood cell.
The patient was transferred to the intensive care unit given worsening multiorgan involvement. Hematology was consulted, and the patient received a simple blood transfusion, fresh frozen plasma, vitamin K, and n-acetylcysteine for progressive hepatic dysfunction. On hospital day 4, laboratory test results were concerning for worsening hepatic function (AST 8,503, ALT 3,340, total bilirubin 20.5, direct bilirubin 9.2, and INR 2.0) and renal function (Cr 5.74). At this point, the decision was made to pursue red blood cell exchange transfusion (Red blood cell exchange [RBCEx]) and hemodialysis. Gastroenterology was consulted for acute liver failure with no preexisting liver disease. After ruling out other etiologies, including drug toxicity, viral or autoimmune hepatitis, Wilson disease, and alpha-1-antitrypsin deficiency, SCIC was the leading diagnosis. In addition to RBCEx, the patient received lactulose and rifaximin for accompanying encephalopathy. Noncontrast brain computed tomography was normal.
By day 5, the patient's hepatic function studies, abdominal pain, and mentation dramatically improved (AST of 3,190, ALT of 2,410, and INR 1.6). These values continued to normalize throughout admission, apart from bilirubin that peaked on hospital day 8 (Figure 1). His clinical status gradually improved to baseline. On follow-up with the patient 6 months later, the patient demonstrated no further sequelae of hepatic dysfunction.
 Figure 1.:
Figure 1.: Visualization of the AST and ALT trend throughout the hospital course with exchange transfusion on hospital day 4, as indicated by the arrow. ALT, alanine transaminase; AST, aspartate transaminase.
DISCUSSIONOur case highlights a successful treatment approach to a rare although serious complication of SCD. Acute liver failure in SCIC has not been well described in the literature. Moreover, given the wide range of presenting symptoms, from benign hyperbilirubinemia to overt multiorgan failure, timely diagnosis and intervention can be challenging.11
Our patient's condition rapidly deteriorated by hospital day 3 with features consistent with previously described cases of SCIC, including marked transaminase elevation, sonographic evidence of cholestasis, hyperbilirubinemia, encephalopathy, and concomitant renal failure. Of note, other cases have described a delayed progression of symptoms, peaking as late as 10 days after admission.1 This variation in timing underscores the importance of maintaining clinical suspicion for hepatic involvement and promptly escalating management as needed.
There is no clear consensus on appropriate management of SCIC, which can include supportive care, simple transfusion, RBCEx, and/or liver transplant. In resource-limited healthcare systems, simple transfusions are often used.1 However, simple transfusions have a relatively higher mortality (88%) and decreased efficacy, as compared with RBCEx in SCIC.1,2,4,7,8,10 RBCEx decreases the fraction of hemoglobin S (HbS), subsequently decreasing viscosity and disrupting vaso-occlusion within the sinusoids.4 In severe cases of SCIC, liver transplant may be considered. However, transplantation poses unique perioperative and postoperative risks with mortality rates that remain high.4,7
In our patient, simple transfusion was attempted first with no clinical response. After RBCEx, hepatic function rapidly improved within 24 hours, consistent with the literature suggesting initial improvement occurring within 48 hours, with complete return to baseline within 6 days.3,12 Other cases have described similar successful outcomes, suggesting that RBCEx should be considered as routine management for acute liver failure in SCIC over simple transfusions.1 Of note, there is conflicting data on optimal post-transfusion HbS goal, with some suggesting <30 mg/DL and others <20 mg/DL.4,13 In our patient, HbS was noted to be 39 mg/DL immediately after one session of RBCEx and 35 mg/DL 2 days later. Given his rapid clinical improvement, the decision was made not to pursue additional exchange to decrease HbS to a predetermined goal.
Our patient exhibited multiorgan involvement, with renal failure requiring temporary hemodialysis. However, improvement in renal impairment without hemodialysis with correction of hepatic dysfunction has also been described.6 Therefore, the necessity and timing of hemodialysis in cases of SCIC with hepatic and renal failure is not well established.
Prior studies have noted that baseline hepatic function and age appear to be predictors of responsiveness to RBCEx in SCIC and should be considered during emergent management decisions.1,8,10 For example, Costa et al described a young patient with SCIC and prior hepatitis C-related cirrhosis that did not improve with RBCEx while others describe similarly poor responses among geriatric patients with underlying liver disease.1,8 Our patient was able to recover after RBCEx without any sequelae, most likely due to his age and otherwise normal baseline liver function.10
The clinical course of SCIC can be unpredictable. Therefore, it is important to consider more aggressive management strategies, such as early liver transplant referrals, especially in patients with underlying liver disease and rapid clinical decompensation. However, transplantation does not address the underlying etiology and has accompanying risks, including graft failure, and often requires subsequent transfusions.6
Owing to the low prevalence of SCIC and lack of diagnostic guidelines, there is limited information on optimal management of SCIC, despite the overwhelming risk of mortality. Our case highlights the importance of early clinical suspicion for hepatic involvement and the utility of emergent and prompt RBCEx, especially in patients without underlying liver disease. Currently, most patients are managed on a case-by-case basis. However, in the future, clinicians may benefit from defining more specific diagnostic parameters and establishing thresholds for escalating care for SCIC.
DISCLOSURESAuthor contributions: Conception, design: N. Wadhavkar, JP Nsubuga. Initial draft: N. Wadhavkar, JP Nsubuga, N. Ibrahim, P. Kumar. Literature review: N. Wadhavkar, N. Ibrahim, P. Kumar. All authors revised the manuscript and approved the final version. N. Wadhavkar is the article guarantor.
Financial disclosure: None to report.
Previous presentation: Poster presented at ACG Annual Scientific Meeting & Postgraduate Court 2023 in Vancouver, Canada.
Informed consent was obtained for this case report.
REFERENCES 1. Adkins BD, Savani BN, Booth GS. Management of sickle cell intrahepatic cholestasis: An argument in favor of automated exchange transfusion. Clin Hematol Int. 2019;1(3):127–33. 2. Likhtshteyn M, Iqbal S, I McFarlane S, Thor S.Intrahepatic cholestasis in a sickle cell patient unresponsive to exchange blood transfusion. Am J Med Case Rep. 2020;7(4):67–70. 3. Brunetta DM, Silva-Pinto AC, do Carmo Favarin de Macedo M, et al. Intrahepatic cholestasis in sickle cell disease: A case report. Anemia. 2010;2011:975731. 4. Khan A, Nashed B, Issa M, Khan MZ. Sickle cell intrahepatic cholestasis: Extremely rare but fatal complication of sickle cell disease. Cureus. 2022;14(2):e22050. 5. Ahn H, Li CS, Wang W. Sickle cell hepatopathy: Clinical presentation, treatment, and outcome in pediatric and adult patients. Pediatr Blood Cancer. 2005;45(2):184–90. 6. Shah R, Taborda C, Chawla S. Acute and chronic hepatobiliary manifestations of sickle cell disease: A review. World J Gastrointest Pathophysiol. 2017;8(3):108–16. 7. Chou ST, Fasano RM. Management of patients with sickle cell disease using transfusion therapy: Guidelines and complications. Hematol Oncol Clin North Am. 2016;30(3):591–608. 8. Schwartz J, Padmanabhan A, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the writing committee of the American society for apheresis: The seventh special issue. J Clin Apher. 2016;31(3):149–62. 9. Stravitz RT, Lee WM. Acute liver failure. Lancet. 2019;394(10201):869–81. 10. Costa DB, Miksad RA, Buff MS, Wang Y, Dezube BJ. Case of fatal sickle cell intrahepatic cholestasis despite use of exchange transfusion in an African-American patient. J Natl Med Assoc. 2006;98(7):1183–7. 11. Praharaj DL, Anand AC. Sickle hepatopathy. J Clin Exp Hepatol. 2021;11(1):82–96. 12. Sheehy TW, Law DE, Wade BH. Exchange transfusion for sickle cell intrahepatic cholestasis. Arch Intern Med. 1980;140(10):1364–6. 13. Howard J. Sickle cell disease: When and how to transfuse. Hematol Am Soc Hematol Educ Program. 2016;2016(1):625–31.
留言 (0)