The data generated in this study are available upon request from the corresponding author.
Reagents
64Cu (t1/2 = 12.7 h, β+; 17.8%, Eβ+max = 656 keV, β−, 38.4%, Eβ -max = 573 keV) was produced on a CS-15 biomedical cyclotron at Washington University School of Medicine (WUSM) with the average specific activity of 343 mCi/μg. Fluorenylmethyloxycarbonyl (Fmoc) amino acids and Rink Amide Resin were purchased from AAPPTec (Louisville, KY, USA). Dichloromethane (DCM), acetic acid, acetic anhydride, thioanisole, phenol, hydroxybenzotriazole (HOBt), HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate), N,N-diisopropylethylamine (DIEA), N-trityl-1,2-ethanediamine, phenol, thioanisol, dimethylformamide (DMF), N,N'-diisopropylcarbodiimide (DIC), trifluoroacetic acid (TFA), iodine, methyl tert-butyl ether (MTBE) and Chelex® 100 sodium form were purchased from Sigma-Aldrich (St. Louis, MO, USA). NODAGA-tris (t-Bu ester) was purchased from Macrocyclics, Inc (Plano, TX, USA). Milli-Q water was obtained from a Millipore Q3 system. All the acetate buffers were prepared with Chelex®-treated milli-Q water. All other chemicals were purchased from Sigma Aldrich unless otherwise noted.
Phage display screening and selection of CD38-specific peptides
Using phage display and purified human CD38 protein (hCD38), the first generation high- and low-selectivity CD38-targeted peptides were identified for the development of CD38-targeted imaging agents [17]. Briefly, two rounds of phage display screening using the PHASTpep platform identified selective peptides for human CD38. CD38 (R&D, Catalog 2404-AC-010, Minneapolis, MN, U.S.A.) or negative control proteins, human CD4 (hCD4), human CD8 (hCD8), and GST-6His were adsorbed on Maxisorp plates (Nalgene, Nunc #442,404) according to NEB phage display protocols (NEB, Ipswich, MA). A 10 µL aliquot of PhD7 library (7 amino acid linear library) (2 × 1011 phage) (NEB, #E8211S) diluted in blocking buffer (DPBS/1% bovine serum albumin (BSA)) was added to each well. After 1 h of incubation at room temperature, wells were washed five times with blocking buffer and the remaining bound phage eluted into 100 μL of glycine buffer (0.2 M glycine, 0.5 M NaCl, pH 2.2) for 9 min before immediately neutralizing with 17 μL of 1 M Tris–HCl (pH 9.2).
For analysis, sequencing of the selected phage clones was performed as described in Brinton et al. [18]. Samples sent to the UVA Biomolecular Research Core Facility were deep sequenced on an Illumina MiSeq Sequencer. FASTQ files were analyzed using PHASTpep [18]. PHASTpep analyzed files were aligned for amino acid sequence homology using the Smith—Waterman algorithm.
Generation of CD38-targeted peptides
Novel CD38 peptide sequences identified using phage display were synthesized via standard Fmoc chemistry [19]. Briefly, using the conjugated peptide NODAGA-PEG4-Thr-His-Tyr-Pro-Ile-Val-Ile-Gly-Gly-Ser-Lys-NH2 (NODAGA-PEG4-SL022-GGS) as an example, the following protocol was used for all peptides synthesized and subsequent bioconjugation in this study. The NODAGA-PEG4-SL022-GGS peptide was prepared using a CEM Liberty Blue microwave peptide synthesizer (Matthews, NC, USA) on Rink Amide resin. The resin (0.1 mmol) was swelled in DCM for 1 h before use. Fmoc amino acids (0.5 mmol, 5 eq), coupling reagent (HBTU, 0.5 mmol, 5 eq), and DIEA (1 mmol, 10 eq) were added to the resin and the mixture was reacted for 15 min under microwave irradiation (100W, 90 °C). The resin was washed three times with DMF. Deprotection of the Fmoc group was carried out by treatment of the resin with a solution of 20% piperidine/DMF for 5 min under microwave irradiation (100W, 90 °C). Subsequently, NODAGA-tris(t-Bu ester) (3 eq) was conjugated to the peptide while still on solid support in the presence of HOBT (5 eq), HBTU (5 eq), and DIEA (6 eq) in DMF to produce the NODAGA-PEG4-SL022-GGS peptidyl resin. The peptide was released from the resin using a cleavage cocktail of TFA: thioanisol: phenol: water (85:5:5:5, v/v/v/v) for 90 min at room temperature. The cleaved peptide product was concentrated in vacuo before performing reverse-phase HPLC purification (Gilson, Middleton, WI, USA). The final product’s NODAGA-PEG4-SL022-GGS (1775 Da) molecular weight was confirmed via electrospray ionization mass spectrometry with peaks observed at 889 (M + 2/2) and 593 (M + 3/3). All other peptide conjugates were synthesized, purified, and characterized in an equivalent manner.
Radiolabeling of NODAGA-PEG4-SL022-GGS with the positron emitter 64Cu
64Cu chloride (64CuCl2) (5 − 10 µL in 0.5 M HCl) was diluted with 0.1 M ammonium acetate buffer (pH 5.5, 50 − 100 µL). NODAGA-PEG4-SL022-GGS (20 µg, 11.3 nmol) was dissolved in 200 µL ammonium acetate buffer (pH 5.5) to which buffered 64Cu solution (37 MBq, 1 mCi) was subsequently added. The reaction mixture was agitated during incubation at 70 °C for 15 min with slight shaking. The radiochemical purity (RCP) of the 64Cu-labeled NODAGA-PEG4-SL022-GGS (64Cu-NODAGA-PEG4-SL022-GGS) was confirmed by analytical radio-reversed phase high performance liquid chromatography (HPLC). Radiochemical reaction progress and purity were monitored using analytical radio-HPLC performed on an Agilent 1260 Infinity (Agilent Technologies, Santa Clara, CA) with a LabLogic Flow-Ram Radio HPLC detector (Lablogic Systems, Sheffield, UK). Finally, a Kinetex 5 µm XB-C18 100A 150 × 4.6 mm LC column was used, and the peptide was eluted with a gradient from 95:5 0.1% TFA in water: 0.1% TFA in CH3CN to 10∶90 0.1% TFA in Water:0.1% TFA in CH3CN over the course of 10 min. Radioactive samples were counted using a Beckman 8000 automated well-type gamma-counter (Beckman Coulter, Franklin Lakes, NJ).
Generation of human MM.1S CD38 knockout cells
CD38 knockout (KO) cells were generated by the Genome Engineering & Stem Cell Center (GESC@MGI) at Washington University in St. Louis. Briefly, synthetic gRNA targeting the sequence 5’- CATCCTGAGATGAGGTGGGTNGG was purchased from IDT, complexed with Cas9 recombinant protein, and transfected into the MM.1S-CBR-GFP cells. Transfected cells were sorted into 96-well plates, and clones were identified using NGS to analyze out-of-frame indels for KO.
CD38 expressing and CD38 knockout human myeloma MM.1S cell line culture
Human myeloma MM.1S cells were initially obtained from American Type Culture Collection (ATCC, Manassas, VA). MM.1S cells were modified to express Click Beetle Red (CBR) luciferase and Green Fluorescent Protein (GFP) (MM.1S-CBR-GFP) by the DiPersio laboratory (Professor John F. DiPersio, Department of Medicine, WUSM, St Louis, USA) in 2014. The expression of CD38 on the MM.1S-CBR-GFP cells (MM.1S-CBR-GFP-WT) was previously confirmed by our lab with ex vivo flow cytometry. Cells tested negative for mycoplasma and were analyzed by the Washington University Genome Engineering and induced Pluripotent Stem Cell Core via MycoAlert PLUS Mycoplasma Detection Kit (Lonza, Visp, Switzerland) in 2018 and 2021, and via MycoStrip Mycoplasma Detection Kit (InvivoGen, San Diego, CA) in 2022. Cell lines were passaged 4–5 times after thawing before use in in vitro and in vivo studies. Cells were routinely cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (ThermoFisher Scientific, Waltham, MA) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Sigma—Aldrich, St. Louis, MO) and 1% penicillin/streptomycin (Gibco, Pittsburgh, PA) at 37 °C with 5% CO2 in a humidified environment. CD38-knockout human myeloma MM.1S clones (MM.1S-CBR-GFP-KO) were developed and validated by the Genome Engineering and Stem Cell Center (GESC) at WUSM. Cell culture conditions for the MM.1S-CBR-GFP-KO cell line were consistent with those of MM.1S-CBR-GFP-WT cell line.
Western immunoblot assays
Cells were mechanically dissociated from tissue culture flasks using rubber-tipped cell scrapers (Sarstedt, Newton, NC) and washed three times with DPBS. Cells were incubated with 25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% Sodium Dodecyl Sulfate (SDS) buffer (RIPA lysis buffer, Abcam, Cambridge, MA) supplemented with EDTA-free (ethylenediaminetetraacetic acid disodium salt hydrate) protease inhibitor cocktail (Pierce, ThermoFisher, Waltham, MA) for 60 min at 4 °C followed by centrifugation at 12,000 × g for 10 min. Protein content was quantified using the bicinchoninic acid protein assay (BCA, Pierce, ThermoFisher Scientific, Waltham, MA). Lysates (10–15 µg total protein) were denatured for 5 min at 100 °C and proteins were separated by non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 100 V for 1.5 h using Mini-PROTEAN TG precast gels with Tris/Glycine/SDS buffer system (Bio-Rad, Hercules, CA). Proteins were transferred to a polyvinylidene fluoride membrane (PVDF, Millipore, Burlington, MA) at 50 V for 2 h. After blocking for 1 h in Tris buffered saline solution supplemented with 0.05% Tween-20 and 3% BSA (TBST blocking buffer, 10 mM Tris–HCl, 150 mM NaCl, and 0.05% [v/v] Tween-20, pH 7.5, 5% (w/v) non-fat milk or 3% BSA (w/v)), the PVDF membrane was rinsed once with TBST and incubated overnight at 4 °C with 1° antibody diluted in TBST blocking buffer. After two 5-min washes, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies in TBST blocking buffer for 1 h at room temperature. Unbound secondary antibodies was removed with three washes in TBST, 10 min each. Bound secondary antibodies were detected using SuperSignal™ West Pico Plus Detection Substrate (ThermoFischer Scientific, Waltham, MA) and ChemiDoc™ Imager (Bio-Rad, Hercules, CA). Primary antibodies used for immunoblotting were rabbit monoclonal anti-CD38 (1:1000, unconjugated, clone EPR4106, Abcam, Waltham, MA) and rabbit monoclonal anti-ß-Actin (1:1000, unconjugated, clone 13ES, Cell Signaling Technologies, Boston, MA). The secondary antibody used in this study was anti-rabbit IgG-HRP (1:10,000, conjugated to HRP, sc-2357, Santa Cruz Biotechnology, Dallas, TX).
Microscale thermophoresis
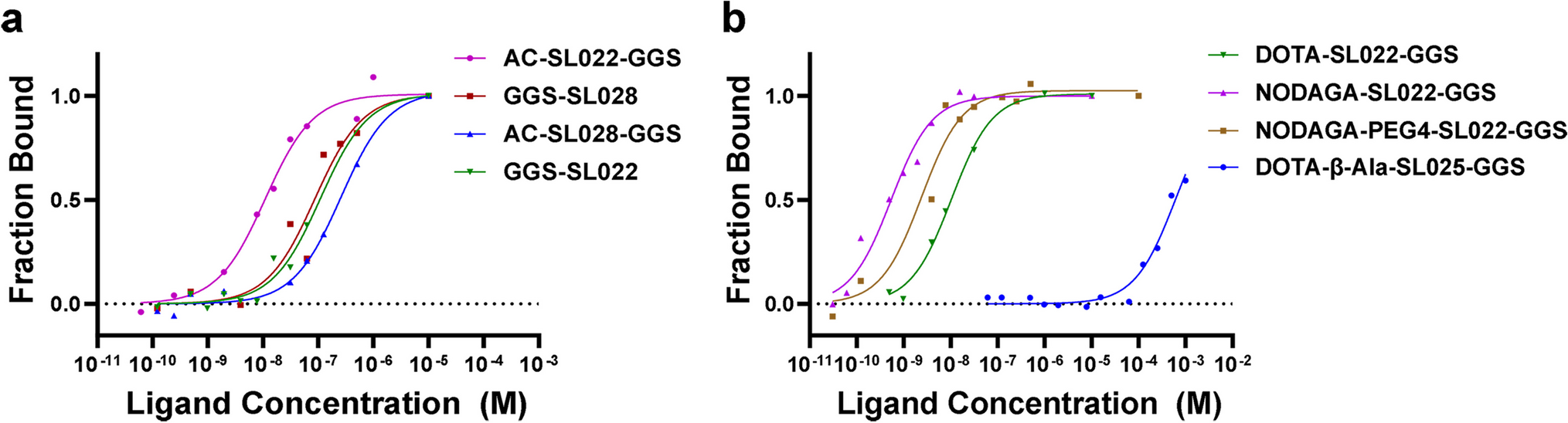
Microscale thermophoresis (MST) assays determined the binding affinity of synthesized peptides to purified CD38 protein and was performed using the Monolith NT.115 (Nanotemper Technologies, Munich, Germany). Respective peptide stock samples were diluted in MST-1 buffer (50 mM Tris–HCl, pH 7.4; 150 mM NaCl; 10 mM MgCl2; 2% DMSO; 0.1% Tween-20; 4% SDS, 4 mM Dithiothreitol) at concentrations ranging from 0.0305 nM to 1000 nM or from 0.0305 µM to 1000 µM. Recombinant human CD38 protein (R&D, Catalog 2404-AC-010, Minneapolis, MN) was labeled with NT647 (Protein labeling kit red-maleimide, Nanotemper Technologies, Germany) and subsequently diluted to 5 nM in MST-2 buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 2% DMSO, and 0.1% Tween-20). Fluorescence was assessed in MST-2 buffer. Samples were loaded into NT.115 standard capillaries, and analysis was performed at 37 °C, 10% LED power, and 40% MST power. The Kd values were calculated from fragment concentration-dependent changes in normalized fluorescence (Fraction Bound) of different CD38 peptides based on the law of mass action using the MO Affinity Analysis v2.3 software.
Flow cytometry
MM.1S-CBR-GFP-WT and MM.1S-CBR-GFP-KO cells were suspended in running buffer (DPBS with 2 mM EDTA and 0.5% BSA). Cell suspension was incubated with fluorochrome-conjugated monoclonal antibody PE/Cy7 CD38 (HIT2; BioLegend, San Diego, CA) in the dark at room temperature for 20 min. Cells were washed once with running buffer, and 7-amino-actinomycin D (7-AAD) was added to stain dead cells. Samples were run on an Attune Cytometer (ThermoFisher Scientific, Waltham, MA) and analyzed using FlowJo V10 (Tree Star, Ashland, OR), and dead cells excluded by gating out 7-AAD positive cells.
Cell binding studies
To evaluate the specificity of the CD38 binding peptide, MM.1S-CBR-GFP-WT cells were suspended in binding buffer (Hank’s Balanced Salt Solution with 1 mM Ca2+ and 1 mM Mg2+ (HBSS + / + , Gibco, ThermoFisher, Waltham, MA) supplemented with 0.1% BSA) at 4 × 105 cells per 250 µL, representing the reaction volume. The cells were incubated with 300 nM 64Cu-NODAGA-PEG4-SL022-GGS (3.3 MBq/nmol) in the presence of 75 µM unlabeled NODAGA-PEG4-SL022-GGS, 75 µM daratumumab, or 75 µM isatuximab at 4 °C for 60 min. The unbound 64Cu-NODAGA-PEG4-SL022-GGS was removed by three washes with HBSS + / + and cell associated activity was determined with WIZARD2® 2480 Automatic Gamma Counter (Perkin Elmer, Waltham, MA).
Animal models
All animal studies were performed in accordance with the WUSM Institutional Animal and Use Committee guidelines. Mice were anesthetized for all injections, treatments, and imaging with 2% isoflurane/100% O2. Ten 5- to 6-week-old Fox Chase Severe Combined Immunodeficient (scid) beige (Charles Rivers Laboratories, Wilmington, MA, USA) or NOD scid gamma (NSG) (The Jackson Laboratory, Bar Harbor, ME, USA) mice were injected with 1 × 106 MM.1S-CBR-GFP-WT cells, described above, in 100 μL DPBS subcutaneously or intravenously via lateral tail vein. Five additional 5- to 6-week-old NSG or Fox Chase scid beige tumor-naive mice served as non-tumor imaging and tissue biodistribution controls. Tumor burden was monitored weekly via bioluminescence imaging (BLI). Mice were randomized into cohorts for small animal PET and tissue biodistribution following administration of the radiolabeled CD38-targeted peptide. The MM.1S-CBR-GFP-WT cell line utilized here was confirmed to have the expected CD38 expression via immunocytochemistry. Tumor burden in mouse tissues was validated in vivo with BLI and ex vivo via histology and immunohistochemistry [20].
In vivo bioluminescence and fluorescence imaging
In vivo BLI was performed by the Molecular Imaging Center at Washington University in St. Louis Medical School on the days indicated on an IVIS 50 (PerkinElmer, Waltham, MA) with Living Image 4.2, a 1–300 s exposure, binning 2–8, FOV 12.5 cm, f/stop1, open filter. Mice were injected intraperitoneally with D-luciferin (150 mg/kg in DPBS, Gold Biotechnology, St. Louis, MO) and imaged under isoflurane anesthesia (2% vaporized in O2). Total photon flux (photons/sec) was measured from fixed regions of interest (ROIs) over the entire mouse using Living Image 2.6. In vivo fluorescence imaging of GFP and SL022-GGS-LS288 (H-THYPIVI1-GGS-K(LS288)-NH2) (Fig. S2) tumor uptake 24 h after intravenous injection was performed on an Optix MX3 time-domain diffuse optical imaging system (Advanced Research Technologies, Montreal, Canada) (Fig. S3). Hair was removed prior to imaging with depilatory cream to improve transmission and imaged using isoflurane anesthesia (2% vaporized in O2). The images were corrected for background to minimize auto fluorescence and diet related non-specific fluorescence.
Histologic assessment
After PET and biodistributions were concluded, the tibia, femurs, and spleen were harvested and fixed for 24–48 h in 10% neutral buffered formalin (NBF). Subsequent bone decalcification was performed with 14% EDTA, pH 7.4 in DPBS for 11–14 days until adequately decalcified. Specimens were processed, paraffin embedded, and sectioned at 5 µm thickness at the Musculoskeletal Histology and Morphometry Core at WUSM, Department of Orthopedic Surgery. Half of the paraffin bone and spleen sections were reserved for immunohistochemical (IHC) analysis, remaining sections were hematoxylin (Surgipath, Richmond, IL) and eosin (Richard-Allen Scientific, Kalamazoo, MI) (H&E) stained using standard procedures for routine histological analysis. Slides were viewed by a pathologist under experimentally blinded conditions.
Immunocytochemistry
Immunocytochemistry of MM.1S-CBR-GFP-WT cells confirmed their positive CD38- and CD31- expression. 10,000 MM.1S-CBR-GFP-WT cells/mL were seeded in Falcon 8-chamber tissue culture treated glass slides (Corning, Tewksbury, MA) and incubated in Phenol-Free Dulbecco's Modified Eagle Medium (DMEM) (Gibco, ThermoFisher Scientific, Waltham, MA) supplemented with L-glutamine (Gibco, ThermoFisher Scientific, Waltham, MA) until confluent. Cells were fixed using ice cold 4% paraformaldehyde in DPBS, pH 7.4 for 10 min at room temperature, washed three times with DPBS, and permeabilized by incubating for 10 min with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in DPBS. Nonspecific binding was blocked by incubating cells with 1% BSA, 0.1% Tween 20 in DPBS for 30 min. After overnight incubation at 4 °C with anti-CD38 mouse polyclonal antibody (# PA5-95,840, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:100 dilution) or anti-CD31 mouse monoclonal antibody (JC/70A #MA5-13,188, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:50 dilution), cells were washed and incubated for 1 h at room temperature with Alexa Fluor 594- or 647-conjugated secondary goat anti-mouse IgG1 (cross-adsorbed, # A-21240, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:50 dilution) or F(ab')2-goat anti-rabbit IgG (H + L) (cross adsorbed, # A48285, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:100 dilution). Cells were subsequently counter stained for nuclei visualization in 0.1 µg/mL Hoechst 33,345 (ThermoFisher Scientific, Waltham, MA) for 1 min and coverslips mounted with fluorescence preserving ProLong Glass Anti-Fade Mountant (Invitrogen, ThermoFisher Scientific, Waltham, MA).
Immunohistochemistry of CD38 expression
Immunohistochemistry (IHC) of the Formalin-Fixed, Paraffin Embedded (FFPE) tumor sections was performed by baking slides for 2 h at 60 °C, deparaffinizing with xylenes, and rehydrating through a graded ethanol to distilled water series. Heat-induced antigen retrieval was carried out using citrate-based Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Tissue was permeabilized and nonspecific binding blocked using 0.2% Triton X-100 (Sigma-Aldrich, St. Louis, MO) and 8% normal goat serum (Sigma-Aldrich, St. Louis, MO) in DPBS. Slides were incubated overnight at 4 °C with anti-CD31 mouse monoclonal antibody (JC/70A #MA5-13,188, Invitrogen, 1:100 dilution) or anti-CD38 rabbit monoclonal antibody (EPR4106 ab108403, Abcam, Waltham, MA, 1:500 dilution) in 3% BSA in DPBS. After washing, Alexa Fluor 647- or 594-conjugated secondary antibodies goat anti-mouse IgG1 (cross adsorbed, # A-21240, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:100 dilution) or F(ab')2 goat anti-rabbit IgG (H + L) (cross adsorbed, # A48285, Invitrogen, ThermoFisher Scientific, Waltham, MA, 1:100 dilution) were added to the slides and incubated for 2 h in the dark. The slides were counter-stained with Hoechst 33,342 (ThermoFisher Scientific, Waltham, MA), and sealed as in ICC protocol.
Microscopic analysis of ICC and IHC
Microscopy of ICC and IHC slides was performed on either Olympus BX51 epifluorescence microscope or an Olympus Fluoview 1000 confocal microscope (Olympus, Center Valley, PA). The Olympus BX51 epifluorescence microscope was equipped with 40x, 60x, 100 × objectives and DAPI, GFP, Texas Red, Cy5, Cy7, and 775/845 filter sets; the confocal microscope was equipped with 505, 560, 635, 705, and 785 nm lasers with 20x, 40x, 60 × objectives. Tumor images were acquired with Olympus cellSens or Cell Standard 1.6 software and exported for analysis with ImageJ software (NIH, Bethesda, MD).
Small animal PET
PET studies were conducted at the Preclinical Imaging Facility at the Mallinckrodt Institute of Radiology, WUSM. Animals from the Fox Chase scid beige (5–6 weeks old males, Charles River Laboratory, Wilmington, MA) or the NSG (5–6 weeks old males, NOD scid gamma, Jackson Laboratory, Bar Harbor, ME) mouse strains bearing either systemically administered MM1.S-CBR-GFP-WT xenograft, a subcutaneous MM1.S-CBR-GFP-WT xenograft, or a tumor-naive control were anaesthetized under continuous Isoflurane vapor (Pivetal®, DCM, WUSM, 2% vaporized in O2) and injected with 6.6 MBq/nmol (1 µg) 64Cu-NODAGA-PEG4-SL022-GGS via lateral tail vein. Imaging was performed 1 h, 2 h, or 4 h after the injection of the indicated bioconjugates using a Mediso nanoScan PET122S/CT1520 in vivo imager (Mediso, Arlington, VA) or Siemens Inveon MM PET/CT scanner (Siemens Corporation, Washington, D.C.). Mediso coincident photons were filtered with an energy window between 400 and 600 keV, and reconstruction was performed with four full iterations, six subsets per iteration with an isotropic voxel size of 0.4 mm3 using the TeraTomo 3D reconstruction algorithm. CT-based attenuation correction was applied for PET reconstruction. Siemens Inveon coincidences were filtered with energy window between 350–650 keV; the reconstruction algorithm was OSEM3D/MAP with 0.8 mm3 voxel size and CT-based attenuation correction.
Tissue biodistribution of 64Cu-NODAGA-PEG4-SL022-GGS
Biodistribution studies were conducted according to previously published protocols [21, 22]. Briefly, 6.6 MBq/nmol 64Cu-NODAGA-PEG4-SL022-GGS was injected via the tail vein. Animals were sacrificed at selected time points after, or after the conclusion of PET, organs of interest were harvested, weighed, and the associated radioactivity determined on a γ-counter. After correcting for background and decay, the percent-injected dose per gram (%ID/g) was calculated by comparison to a weighed and counted standard.
Secondary analysis of Multiple Myeloma Research Foundation (MMRF) CoMMpass study data
We performed a secondary analysis of data from the MMRF CoMMpass study (Courtesy, Mark Fiala, PhD and Ravi Vij, MD). CoMMpass is a longitudinal study of over 1000 MM patients which incorporates both clinical and genomic data. As part of the CoMMpass study, RNA-sequencing (RNA-seq) on CD138-enriched bone marrow cells was performed using Illumina TruSeq RNA library kits by Translational Genomics Research Institute (TGEN). For this analysis, data were extracted from the MMRF Researcher Gateway Portal corresponding with Interim Analysis 22.
Statistical analysis
Data was presented as mean ± SD and statistical analysis performed using GraphPad Prism Version 9.1.0 software (GraphPad Software, Inc., La Jolla, CA) and Microsoft Excel. A two-tailed unpaired t-test was used for data with two groups and one variable. Data with one variable and multiple groups were analyzed with a one-way ANOVA and Tukey's or Dunnett's multiple comparisons test to determine the adjusted P-value. Data with two variables and multiple groups were analyzed with a two-way ANOVA and Tukey's multiple comparisons test to determine the adjusted P-value. P values of less than 0.05 were considered statistically significant.




留言 (0)