
記住我
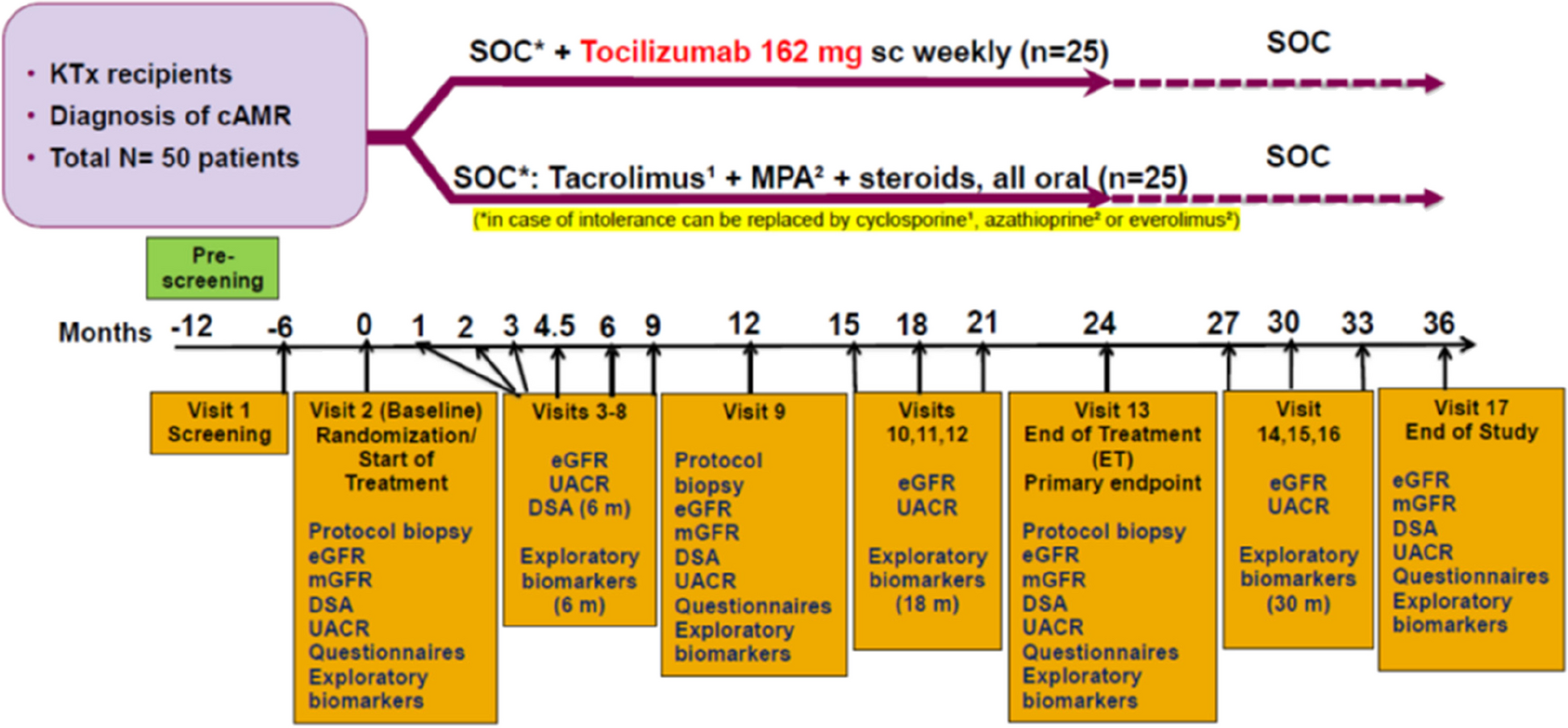







The study is designed as a monocentric, prospective, randomized (parallel group; allocation ratio 1:1), outcome-blinded, controlled interventional trial (exploratory RCT) at the Head and Neck Cancer Center of the University Hospital Regensburg. The intervention consists of individual information and risk consulting, individual logopedic counseling with instructions for exercise treatment of swallowing function, and risk-related nutritional counseling (face-to-face). In the control group, patients are treated according to the currently applicable standard of care described in the guidelines on laryngeal cancer [8] and oral cavity cancer [9] as well as in the survey on Head and Neck Cancer Centers of the German Cancer Society (https://www.krebsgesellschaft.de/zertdokumente.html).
OutcomesAll patients participating in the study are analyzed by means of various questionnaires in addition to the therapy standard of the Head and Neck Cancer Center Regensburg:
EORTC-QLQ-C30 for the general assessment of QoL [10]
EORTC-QLQ-HN43 to assess the QoL of patients with HNSCC [11]
Dysphagia Handicap Index (DHI) for measuring the handicapping effect of dysphagia on the physical, functional, and emotional aspects of people’s lives [12]
Hospital Anxiety and Depression Scale (HADS) for assessing anxiety and depression [13]
Functional Oral Intake Scale (FOIS-G) to analyze oral food intake [14]
In addition, a bioelectrical impedance analysis, a determination of the body mass index (BMI), and measurement of serum albumin level take place.
Furthermore, a detailed phoniatric diagnosis of swallowing (Functional Endoscopic Evaluation of Swallowing (FEES)) regarding restrictions of oral nutrition and aspiration risk, which is objectively evaluated by means of the Penetration-Aspiration Scale (PAS) [15] and the Yale Residue Scale (YRS) [16], is performed.
Primary study endpoints, 6 weeks after the end of therapy:
Swallowing function (objectively assessed using FEES: PAS, YRS, subjectively using DHI and FOIS-G)
Emotional distress: anxiety and depression (HADS)
Secondary study endpoints 6 weeks, 3 and 6 months, and 9 or 12 months after the end of oncologic therapy:
Swallow-related QoL (DHI; EORTC-QLQ-HN43; FOIS-G).
◦ Time to decannulation
◦ Percentage of nutrition by gastric feeding tube/time of dependence on enteral nutritional substitution via a gastric feeding tube
◦ Incidence of complications (aspiration pneumonia)
Inpatient length of stay, incl. readmission
Nutritional status (BMI, albumin, bioelectrical impedance analysis)
General QoL (EORTC-QLQ-C30)
Occupational reintegration
Subgroup analyses regarding older patients (> 65 years) and patients with weaker social status
Eligibility criteria of study participantsInclusion criteria:
Exclusion criteria:
T1 glottic carcinoma, salivary gland tumors, sinus, nasal cavity carcinomas, lip carcinomas, skin carcinomas
Planned laryngectomy, total glossectomy, esophageal dysphagia
No curative therapy
State after therapy of carcinoma of the upper aerodigestive tract or esophageal carcinoma
State after radiotherapy in the head and neck region
Higher-grade cognitive impairment (e.g., dementia, Korsakow syndrome)
Psychomotor impairment (e.g., Parkinson's disease), previous neurological diseases with dysphagia (e.g., post apoplexy)
Age < 18 years
ECOG > 2
Patient is unable to complete questionnaires even with assistance (inadequate ability to read and write, higher grade inadequate hearing loss).
Language barrier
Pregnancy/breastfeeding
InterventionsFor a detailed analysis of the effects of Phoniatric PREhabilitation in Head and Neck Cancer patients on Aspiration and Preservation of Swallowing, two study arms were designed (Table 1). In the intervention group, additional measures take place compared to the control group:
General information and risk counseling on the swallowing examination, also for the prevention of dysphagia before and during therapy with video demonstration [17], if necessary, also to accompanying relatives.
Logopedic exercises to strengthen and improve the sensitivity and range of motion of oral, pharyngeal, and laryngeal muscles (tongue, larynx, jaw, e.g., Mendelsohn maneuver, Shaker maneuver, effortful swallow, supraglottic swallowing technique). The exact therapeutic procedure is based on an individual protocol. The patient is instructed to discontinue or modify in-home exercises (eating rules, individual exercise protocol with recommended dosage, documentation in exercise diary). The prescribed dose of exercise treatment takes into account not only the need but also the expected patient’s adherence to therapy. It includes at least 10 repetitions 3 times a day for each of, e.g., 5 forms of exercises [4], so that a total dose of 150 exercises per day is achieved. Even in the case of minor symptoms, preventive counseling and exercise treatment are carried out with a view to the future. After 3–7 days, a speech therapist conducts a control interview (face-to-face or by telephone) based on the exercise diary. If it seems necessary, an intervention can take place at short notice.
Additional nutritional risk screening [18] and risk-related nutritional counseling (face-to-face), performed by staff of the Department of Internal Medicine III, Hematology and Oncology.
Table 1 Study design—intervention versus control groupSample size and recruitmentSince this type of therapeutic approach was not studied yet in this form, it was not possible to estimate an expected effect size for a sample size calculation. Therefore, the study design for the present project was chosen with a complex family of endpoints to obtain first estimates of treatment effects for the clinically relevant endpoints with the consequence that no sample size calculation based on an expected treatment effect of one primary endpoint was performed. Instead, sample size considerations are based on assumptions about possible effect sizes of different endpoints (primary endpoint family) with the aim to get reliable and valid effect estimates for a following more extensive multicenter clinical trial. After sharpening the focus, the results of this study will be used to initiate a multicenter confirmation study, e.g., within the framework of the BZKF (Bavarian Center for Cancer Research, Head and Neck Tumor Study Group). According to the publication by Guillen-Sola [20] we assume median effect sizes within the primary endpoint family. They state an effect in the Penetration-Aspiration-Scale of 2 Units and a difference ≥ 10 units in the tests of quality of life (EORTC-QLQ-C30 and EORTC-QLQ-HN35). This is also the expectation of the therapy based on our clinical experience. Here, a “median effect size” methodologically means a Cohen’s d of approximately 0.5 or half a standard deviation [21].
To obtain sufficient accuracy for the estimation of treatment effects due to the performed intervention as well as for the planning of a confirmative follow-study, n = 30 patients per study arm (n = 60 in total) are to be analyzed under the assumption of median effect sizes regarding the endpoints of the primary endpoint family [22]. Assuming a drop-out rate of 15%, n = 70 patients have to be included and randomized in the study. In the recruitment period of 17 months, we assume a potential study population of 350 patients (250 primary cases in the Head and Neck Cancer Center per year). Considering the increase in higher tumor stages due to the corona pandemic, we expect 40% of patients (n = 140) to meet all inclusion and exclusion criteria, of which 50% of patients (n = 70) are likely to give informed consent. Thus, inclusion of 70 patients in the planned recruitment period is realistic. Screening and identification of potential study patients will occur during the patient’s first inpatient stay (primary diagnosis, histology acquisition, pan-endoscopy, staging). After the tumor diagnosis has been confirmed, the first contact with the potential study patient is made in the Ear, Nose and Throat Clinic and the Clinic for Oral and Maxillofacial Surgery (usually after the interdisciplinary tumor board) by physicians from the clinics with the support of the study assistance. The detailed study information as well as the study inclusion is carried out by the medical staff of the Section of Phoniatrics and Pediatric Audiology.
The study is explained in detail to the patient by a physician from the Section of Phoniatrics and Pediatric Audiology. Subsequently, the patient gives an informed consent to participate in the study by means of a written declaration of consent. As part of the consent process, the participant will be asked whether the data collected may also be used for questions unrelated to the study but related to the study purpose. The participant can agree to such use or refrain from doing so. Randomization is performed prior to the intervention, after informed and signed consent, via REDCap database. Each randomization is documented and signed by the investigator.
Statistical analysisThe primary outcome measure is a complex family of endpoints including swallowing function (subjectively using DHI, FOIS-G, and objectively using FEES) and anxiety and depression (HADS) 6 weeks after the end of therapy. Each questionnaire score will be calculated using the associated manual. The two study groups will be compared for each endpoint using analysis of covariance (ANCOVA). The respective endpoint at the time point after 6 weeks is included in the model as the dependent variable, the study group as factor, and the respective baseline value at baseline as covariate. To check the endpoints, the intention-to-treat collective will be used as the evaluation collective. Analyses will also be evaluated on the per-protocol collective as a control. Established, clinically relevant effect sizes will be used as a comparison to evaluate the effects (estimated marginal means and 95% confidence intervals) and their benefit to patients. A panel of experts (phoniatrics, ENT, maxillofacial surgery, speech therapy, study assistance, biometry) will evaluate which changes in primary outcome parameters should be interpreted as clinically relevant. In addition, subgroup analyses will be performed regarding older patients (> 65 years) and patients with weaker social status. The concept of complex pre-therapeutic phoniatric prehabilitation is considered promising if there is a consistent advantage over the control group within the complex family of outcomes.
Secondary study endpoints 6 weeks, 3 and 6 months, and 9 and/or 12 months after the end of oncologic therapy will be considered exploratively: swallowing-related QoL, subjectively perceived swallowing function, incidence of aspiration pneumonia, general QoL, nutritional status, time to decannulation, nutritional percentage by gastric feeding tube, time of dependence on enteral nutritional substitution via gastric feeding tube, duration of inpatient stays, and occupational reintegration.
The analysis of secondary endpoints is purely descriptive and exploratory. Depending on the type of endpoint, covariance analyses for metric endpoints, logistic regressions for dichotomous endpoints, or simple tests such as Student’s t-tests, Wilcoxon Mann–Whitney U tests, or chi-square tests are used.
Randomization and blindingRandomizationRandomization of a patient is stratified according to UICC stage (8th edition) into stratum I (UICC I and II) and stratum II (UICC III and IV). This considers that patients treated by surgery alone are generally assigned to the earlier tumor stages UICC I and II and are less likely to require adjuvant therapy. Patients with an indication for adjuvant radiotherapy or radiochemotherapy or primary radiotherapy or simultaneous radiochemotherapy are more likely to belong to the advanced tumor stages UICC III and IV. Randomization will be computerized (REDCap database) by a physician from the Section of Phoniatrics and Pediatric Audiology prior to intervention, after informed and signed consent (see above).
BlindingThe study is designed outcome-blinded, meaning that each detailed phoniatric swallowing examination (FEES) is subsequently assessed again objectively by another physician from the Section of Phoniatrics and Pediatric Audiology based on the video documentation using the specified documentation parameters (PAS, YRS). The second assessor does not know whether the examined patient is from the control or intervention group. Unblinding is not provided.
Trial flowIn the intervention group, the inclusion examination is followed by nutritional counseling and a first control interview by a speech therapist based on the exercise diary taking place in the presence or on the phone after approximately 3–7 days. Further appointments are scheduled at 6 weeks, 3 and 6 months, and 9 or 12 months after the end of oncological therapy (Fig. 1).
Fig. 1
PREHAPS—trial flow chart
There will be no special post-trial care, but the standard surveillance program.
Data managementIn this study, the Research Electronic Data Capture (REDCap) system is used to implement electronic case report forms (eCRFs). All participant data throughout the study will be collected and directly entered into eCRFs. Data entries will be time-stamped and user-tracked, providing a detailed audit trail for monitoring data changes.
The randomization module of REDCap will be used to assign participants to one of both study groups (intervention/control group). The system’s ability to implement complex randomization algorithms facilitated the stratification process, ensuring a balanced allocation of participants across different study groups and strata.
Quality assuranceMonitoringOnsite monitoring follows a risk-based approach and is laid down in a monitoring plan. 100% SDV include informed consent and randomization. Within the established data management process, specific data quality rules are applied to collected data. Any deviation, unexpected missing, or implausibility of data will be reported to the study site for clarification.
Data monitoring committeeThe data monitoring committee (DMC) is a panel of experts (phoniatrics, ENT, maxillofacial surgery, speech therapy, study assistance, biometry) and will evaluate which changes in primary outcome parameters should be interpreted as clinically relevant (see above). Interim analyses are not provided.
留言 (0)