Study Design, Ethical Approval, and Registration
This prospective, double-blind randomized controlled trial took place at the Anhui Provincial Maternal and Child Health Hospital, China, between May and July 2023. The study adhered to the Declaration of Helsinki and received the approval from the hospital Ethics Committee (Approval No. YYLL2023-04-01-01, April 1, 2023). The study protocol was registered in the Chinese Clinical Trial Registry (ChiCTR2300071069, http://www.chictr.org.cn/index.aspx). All participants provided the informed consent.
Research Participants and Inclusion/Exclusion Criteria
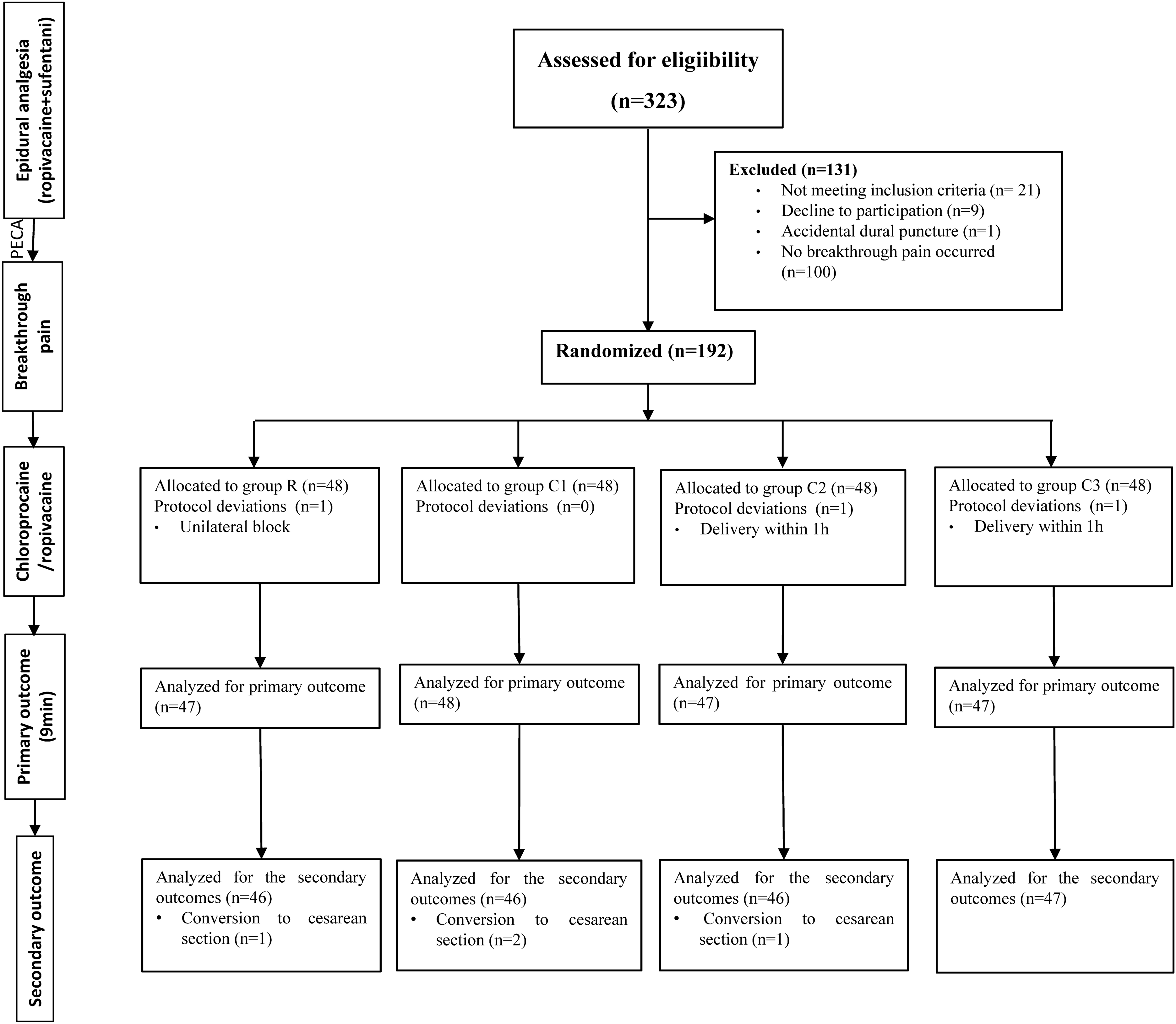
Recruitment occurred after the parturients entered the delivery room before the initiation of labor analgesia. Eligible patients were primiparas with singleton, full-term pregnancy, normal fetal position, aged between 20 and 36 years, a body mass index (BMI) of 21.0–35.0 kg/m2, American Society of Anesthesiology (ASA) physical status I or II, scheduled for vaginal delivery under epidural analgesia, and experienced BTP after epidural analgesia. BTP was defined as a numerical rating scale (NRS) score > 3 after initial pain relief following at least one patient-controlled epidural analgesia (PCEA) bolus administration. Exclusion criteria included patient refusal of additional analgesic, high-risk pregnancies (placental abruption, severe preeclampsia, and placenta previa), known fetal abnormalities, history of chronic pain or prenatal psychotropic drug use, poor mental cooperation, allergies to opioids or local anesthetics, delivery within 1 h post-analgesia, and cases where labor analgesia required an emergency cesarean section.
Randomization and Blinding
A computer-generated random number generator was used to randomize study participants on a 1:1:1:1 ratio to receive different concentrations of local anesthetics for labor analgesia.
Group R (control):
1.5 mL of 0.5% ropivacaine + 4.5 mL of normal saline (0.125% ropivacaine).
Group C1:
1 mL of 3% chloroprocaine + 5 mL of normal saline (0.5% chloroprocaine).
Group C2:
2 mL of 3% chloroprocaine + 4 mL of normal saline (1.0% chloroprocaine).
Group C3:
3 mL of 3% chloroprocaine + 3 mL of normal saline (1.5% chloroprocaine).
The final volume of the study drugs was 6 mL. There were no intravenous drugs administrated perioperatively. A nonparticipating nurse prepared these drugs. Group allocations were concealed from patients, obstetricians, and research staff involved in patient enrollment and data collection. All patients were accompanied by one of the investigators (LH or CZ) throughout the labor. Data were recorded until the end of labor.
Labor Analgesia Protocol
In the labor room, every parturient was monitored by pulse oximetry, noninvasive blood pressure measurement, and fetal tocodynamometry. An intravenous access was established. The parturient was placed in the left lateral position. At the estimated L2–3 interspace, the epidural procedure was performed using a 16-gauge needle and the loss of resistance to air technique. Subsequently, a 19-gauge multi-orifice epidural catheter was advanced approximately 4 cm into the epidural space. After the administration of a test dose of 3 mL of 1.0% lidocaine, epidural analgesia was initiated and maintained using a solution of 0.09% ropivacaine combined with 0.4 µg/mL sufentanil (10 mL). The sensory block level was assessed using an alcohol-soaked cotton swab. If the sensory block level was below T10 after 15 min, an additional 5 mL of the analgesic solution was administered. Thirty minutes later, if the analgesia was deemed insufficient (NRS score > 3), the epidural catheter placement was considered unsuccessful, and the parturient was excluded from the study.
After successful epidural analgesia initiation, the labor analgesia was continued by PCEA. Analgesia via programmed intermittent epidural bolus (PIEB) commenced with a 10-mL bolus 30 min after pump initiation. It was sustained with an hourly 10-mL programmed bolus and complemented by 7-mL PCEA boluses, with a 20-min lockout interval. The hourly epidural volume reached a maximum of 31 mL.
Breakthrough Pain Management Protocol
Patients undergoing epidural labor analgesia were randomly assigned to analgesic treatment with different study drugs once they experienced BTP during labor. Once the BTP occurred, every parturient received the experimental analgesic and was evaluated every 3 min for 15 min using the maternal NRS (0–10, where 0 and 10 indicated no pain and severe pain, respectively) for pain. If the experimental randomized treatment did not provide adequate pain relief, the alternative treatment was administered. Subsequent evaluation occurred 15 min post-administration of the second solution. When the pain score did not decrease by at least 4 points from the baseline 15 min after the second drug administration, 5 mL of 2% epidural lidocaine was administered. If the third dose failed to decrease the visual analog scale pain score by at least 4 points below the baseline within 15 min (45 min post-initial study drug), the epidural catheter was deemed nonfunctional, and the case was excluded from further analysis.
Outcome Measurements
The primary outcome was the treatment’s success rate for BTP. This randomized, double-blind trial aimed to investigate the onset of action and analgesic effect of chloroprocaine compared with ropivacaine in breakthrough pain relief. Unlike a previous study [12], we defined treatment success as a decrease in the NRS score by at least 4 from the baseline during active uterine contraction observed 9 min post-drug administration.
Secondary outcomes included the following: (a) number of patients with effective analgesia (decrease in the pain score by at least 4 from the baseline at 15 min post-administration); (b) maternal NRS pain scores assessed every 3 min for 15 min after injection, with the NRS scores recorded every 15 min for 15–60 min after the initial injection and then every hour until delivery; (c) modified Bromage scores (1 = complete motor block; 2 = almost complete, allowing only foot movement; 3 = partial, allowing knee movement; 4 = detectable hip flexion weakness, allowing leg raise but not sustained; and 5 = no weakness, allowing leg raise for 10 s; 6 = no weakness), evaluated at 15 min post-injection; (d) ropivacaine and sufentanil consumption; and (e) maternal satisfaction gauged at 1 and 24 h post-delivery using a 10-cm visual analog scale.
Additional secondary outcomes encompassed the following: (1) stages leading to delivery; (2) potential adverse effects such as postpartum headache, itching, nausea, and vomiting; (3) hypotension incidence, marked by systolic blood pressure decreasing below 90 mmHg or declining more than 20% from the baseline; (4) outcomes concerning neonates; and (5) obstetric outcomes.
Sample Size Calculation
The sample size was estimated using PASS v11.0 (PASS, NCSS, USA). The primary outcome of this study was the “success” rate of epidural injection analgesia for BTP. In our preliminary study involving four patient groups (10 patients/group), the success rates of epidural analgesia for BTP in groups R, C1, C2, and C3 were 40%, 50%, 60%, and 80%, respectively. With a power of 0.90 and an α level of 0.05, a minimum of 38 participants per group, with a total of 152, were required. Considering a 20% attrition rate, we decided to enroll 48 participants in each group, with a total of 192 participants.
Statistical Analysis
A per-protocol analysis was conducted. Data were presented as either mean ± standard deviation or median (interquartile range) unless stated otherwise. The Kolmogorov–Smirnov test was used to assess the normality of continuous data. Variables were categorized and analyzed on the basis of their standard or skewed distribution. For continuous variables, either analysis of variance (ANOVA) or the Kruskal–Wallis H test was used. Categorical data were presented as proportions and analyzed using the chi-squared or Fisher’s exact test. Odds ratio (OR) and 95% confidence intervals (CI) were reported. The linear mixed model was used to compare pain scores across groups at specified time points, adjusting for baseline scores. A Bonferroni correction accounted for multiple comparisons. The initial experimental randomized drug loading dose had an endpoint recorded at 0 min. Only scores from the first 3 h were included in the analysis. All data were analyzed using an intention-to-treat principle. Statistical significance was set at a two-sided p value of less than 0.05. Statistical analyses were performed in SPSS 26.0 (Chicago, IL, USA).



留言 (0)