
記住我
Atypical hemolytic uremic syndrome (aHUS) is a form of thrombotic microangiopathy (TMA) that results from the defective regulation of the alternative complement pathway. aHUS is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal dysfunction (1–3). Genetic abnormalities associated with complement activation in the alternative pathway occur in 50%–60% of aHUS patients (1).
The complement factor H (FH) has an important role in regulating the alternative pathway and is considered a common genetic factor underlying aHUS (3, 4). With respect to cases of aHUS with complement factor H (CFH) gene mutations, prognosis is poor and affected patients progress to end-stage renal disease if not treated appropriately (5).
The CFH gene along with genes for complement Factor H-related (FHR) proteins 1, 2, 3, 4, and 5, are located within the regulators of complement activation (RCA) cluster on chromosome 1q32 (3, 6). Sequence analyses have revealed numerous duplications in this region because of the high degree of sequence homology. Nonallelic homologous recombination (NAHR) events occur within this repetitive sequence, which result in the formation of a CFH::CFHR fusion gene. The primary mutations in the CFH gene typically occur within the final one to three short consensus repeats (SCRs) (3, 7). Several fusion genes resulting from NAHR in the CFH–CFHR gene region have been identified, including CFH::CFHR1, CFH::CFHR3, and CFHR1::CFH (3, 8–12).
There is some confusion in the nomenclature of the CFH gene. The very small exon (Exon 10) within CFH gene only codes for 4 amino acids, forming the splicing isoform of FHL-1 (13). In this report, we designated the number of exons of the CFH gene as 23 including this very small exon. Three types of CFH::CFHR1 fusion genes have been reported, initially described by Venales et al. (3, 8), where the two last exons of CFH are replaced with exons 5-6 of CFHR1. The second CFH::CFHR1 fusion gene, reported by Maga et al. (3, 9), involves the replacement of exon 6 of CFHR1 starting from the final exon of CFH. The third CFH::CFHR1 fusion gene, reported by Piras et al. (10), involves the replacement of exons 4-6 of CFHR1 starting from the three last exon of CFH. Regarding the combination of a CFH::CFHR1 fusion gene and CFHR gene duplication, while CFH::CFHR1 fusion gene and de novo CFHR1 duplication has been previously reported (10), the simultaneous occurrence of this fusion gene with CFHR large duplication including the CFHR2 and CFHR4 gene has not been previously reported.
Here, we encountered a family with aHUS spanning four generations and identified a novel dual CFH::CFHR1 fusion gene and CFHR gene duplication within one allele of the CFH and CFHR gene region. The fusion gene encompasses exons 1–22 of CFH and exon 6 of CFHR1, similar to the report by Maga et al. (9). However, in our case, this anomaly further included a duplication of the CFHR3-1-4-2 genes. This extensive duplication is the first report in the world.
2 Case presentation of the probandAn 8-month-old girl (proband) initially presented with mild diarrhea that lasted one week. Her activity decreased and she was passing brown urine. After visiting the emergency center, she was admitted to our hospital the following morning. She was born after an uneventful 38-week pregnancy at a birth weight of 2,762 g (−0.18 SD). Her developmental milestones were normal. She appeared pale and there were signs of anemia in the conjunctiva of her eyelids. Petechiae were observed on her lower extremities, but urine volume was normal.
The laboratory results revealed the following: Hemoglobin: 6.5 g/dL, Platelet count: 7.0 × 103/μL, serum creatinine (Cr): 1.27 mg/dL, lactic acid dehydrogenase (LDH): 3,932 mg/dL, total bilirubin: 2.7 mg/dL, Coombs test: negative, haptoglobin (Hp): undetectable, complement components within normal ranges (C3: 78 mg/dL, C4: 33 mg/dL), and negative anti-lipopolysaccharide (LPS)-IgM antibody. A blood smear showed the presence of fragmented red blood cells. Urinalysis indicated proteinuria (Protein/Cr ratio: 85.3 g/gCr) and hematuria. Stool cultures and occult blood tests were negative. Based on these findings, she was diagnosed with aHUS.
Treatment was initiated with a daily plasma exchange on the third day of hospitalization, resulting in the cessation of further increases in serum Cr levels and a reduction in LDH and total bilirubin; however, her platelet count remained low. By the fifth day, Streptococcal TMA, cobalamin C defect-HUS, and Shiga toxin-producing Escherichia coli infection were ruled out. ADAM metallopeptidase with thrombospondin type 1 motif 13 (ADAMTS13) activity was within the normal range.
On the sixth day, because of a positive hemolysis test, she was administered a dose of ravulizumab (Rav) at 600 mg based on her weight. Subsequently, the laboratory results showed improvement. By the thirteenth day, her platelet count had risen to 700 × 103/μL, and her serum Cr level had dropped to 0.38 mg/dL. She continued to receive Rav on a monthly basis and remained disease-free for two years with no signs of recurrence.
3 Family historyHer parents were nonconsanguineous and she did not have any siblings. Both of her parents were healthy and had no history of TMA; however, her paternal grandfather experienced sudden renal failure in his thirties and had been on dialysis for 40 years (Figure 1A). He was initially diagnosed with rapidly progressive glomerulonephritis based on a renal biopsy. Upon reevaluation of his kidney biopsy specimen, which was collected 40 years ago, it was evident that the pathology was consistent with TMA.
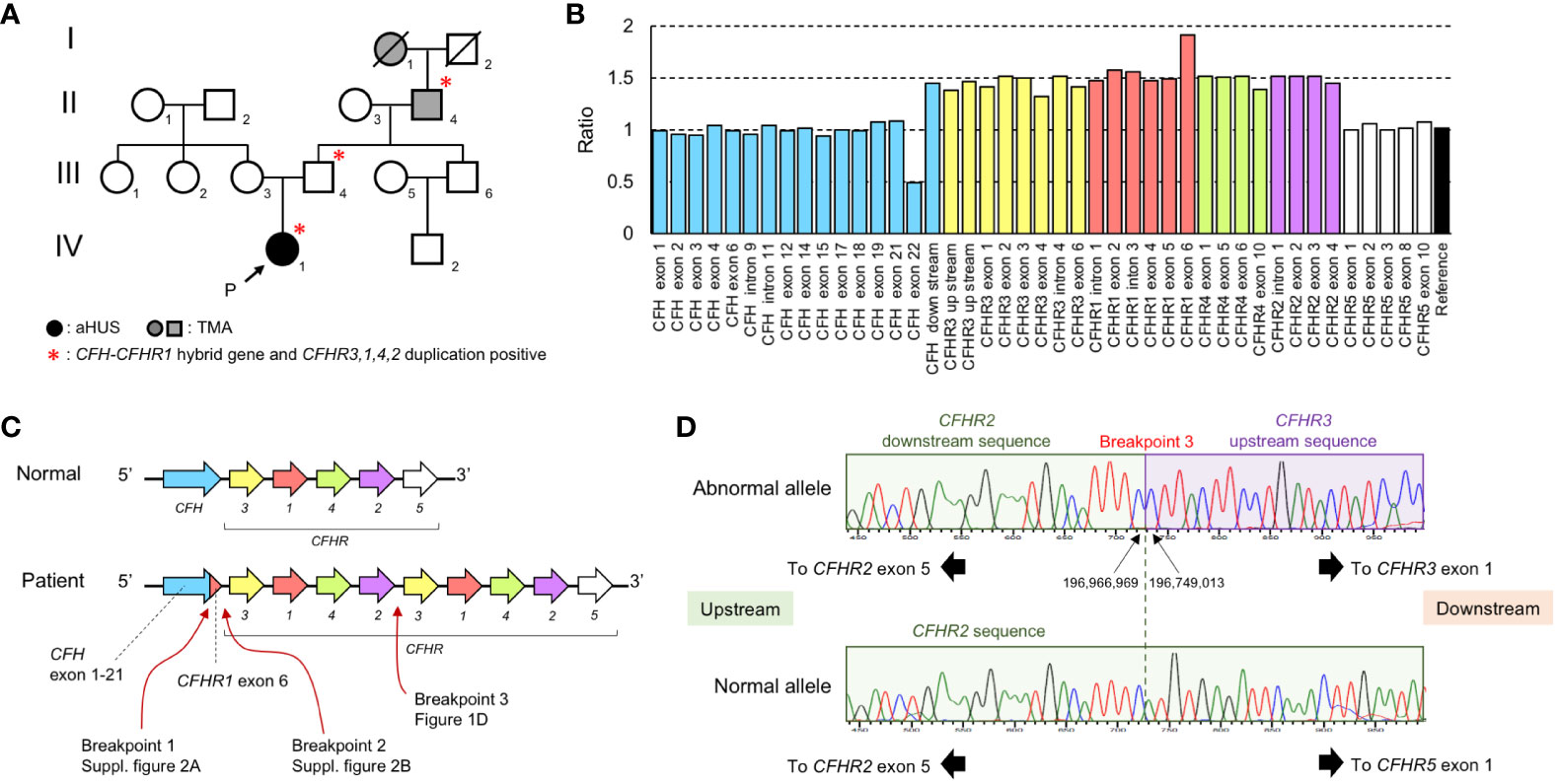
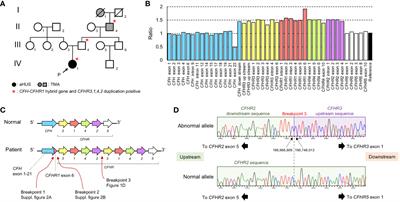
Figure 1 Laboratory and gene analysis of CFH and CFHR. (A) Family tree. There were 3 members of the family who were affected by aHUS or TMA (I-1, II-4, and IV-1) and 3 members of the family (II-4, III-4, and IV-1) carrying the CFH::CFHR1 fusion gene and CFHR3-1-4-2 duplication.I-1: Great grandmother II-4: Grandfather III-3: Mother III-4: Father IV-1: Proband. (B) The result of MLPA analysis in the patient (IV-1). MLPA analysis over the CFH–CFHR region shows a 1.5-fold ratio in a large region beginning with CFH downstream and ending CFHR2 gene exon 4. The numbering of CFH exon in the graph follows the instructions for use of the SALSA® MLPA® Probemix P236-B1 CFH Region. (C) Estimation of CFH–CFHR region of the patient. The CFH::CFHR1 fusion gene is caused by a gene conversion between CFH gene exon 23 and CFHR1 gene exon 6. The duplication occurred from the CFHR3 gene to the CFHR2 gene. (D) Sequencing analysis of breakpoint between CFHR2 and CFHR3 gene (Breakpoint 3). In the normal allele, CFHR5 exon 1 exists after the CFHR2 downstream sequence. CFHR3 exon 1 exists after the CFHR2 downstream sequence in the abnormal allele. The breakpoint is located in a region 2 bp between the CFHR2 downstream and CFHR3 upstream region.
Five years before the proband’s birth, her paternal great grandmother suffered from anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis in her eighties. Her renal pathology indicated necrotizing vasculitis and she was administered steroid treatment. Three months after the onset of ANCA-associated vasculitis, she developed pneumonia and hematologic TMA. The laboratory findings revealed the following: Hemoglobin: 6.5 g/dL, Platelet count: 1.7 × 103/μL, Cr: 5.08 mg/dL, and LDH: 2,376 mg/dL. A blood smear showed the presence of fragmented red blood cells. Her ADAMTS13 activity was within the normal range and haptoglobin was <10%. Testing for O157-LPS yielded negative results. Unfortunately, she passed away without responding to various treatments. Although TMA was considered a possible diagnosis in her case, a definitive diagnosis was never established.
4 Laboratory and genetic analysis of the CFH and CFHR genesBecause TMA was identified in this family over four generations, additional tests were performed. The patient, her father, and paternal grandfather exhibited hemolytic reactions in their citric acid plasma in a hemolysis test using sheep erythrocytes (Supplementary Figure 1) (14). The degree of hemolysis in the citric acid plasma of the mother was similar to that of a healthy control. After the proband received Rav, the degree of hemolysis in her citric acid plasma decreased to that of the healthy control.
Genetic abnormalities in the CFH and CFHR genes were analyzed by Multiplex ligation-dependent probe amplification (MLPA). We conducted the MLPA analysis using the SALSA® MLPA® Probemix P236-B1 CFH Region from MRC Holland (Amsterdam, The Netherlands). The relative copy number ratio of the CFH exon 23 (exon 22 in the Figure 1B) was 0.5, the ratio of the region downstream of CFH to CFHR2 exon 4 (excluding CFHR1 exon 6) was 1.5, and the ratio of the CFHR1 exon 6 was 2.0 (Figure 1B). Because this pattern followed an autosomal dominant inheritance, we hypothesized that a CFH::CFHR1 fusion gene, in which CFH exon 23 was replaced by CFHR1 exon 6, along with a CFHR3-1-4-2 gene duplication, existed on the same allele (Figure 1C).
PCR and direct sequencing were done using primers specific to the CFH intron 22 and CFHR1 exon 6. We confirmed the presence of a CFH::CFHR1 fusion gene (NC_000001.11: g.196,746,919_196,748,226con196,831,605_196,832,909). The rearranged region upstream of the CFH::CFHR1 fusion gene is presented in Supplementary Figure 2A. The rearranged region of downstream of the CFH::CFHR1 fusion gene was also analyzed (Supplementary Figure 2B). Furthermore, we verified that the cytosine 7,347 on the 3′ side of CFHR2 exon 5 was continuous with the cytosine 25,827 on the 5′ end of the CFHR3 exon (Figure 1D). Based on these results, we concluded that the gene sequence of the patient consisted of a CFH exon 1-22::CFHR1 exon 6 fusion gene and CFHR3-1-4-2 gene duplication (NC_000001.11: g.196,749,013_196,966,969dup).
5 DiscussionIn this case study, a proband and her family carried both the CFH::CFHR1 fusion gene and a duplication of the CFHR3-1-4-2 genes. The presence of the CFH::CFHR1 fusion gene resulted in the replacement of the C-terminus of the FH protein with the FHR-1 protein (3, 8–10). This fusion gene encodes a hybrid FH/FHR-1 protein, in which the last SCR20 of the FH is substituted with SCR5 from FHR-1.
FH SCR20 and FHR-1 SCR5 are similar in their structural features and binding properties. Both SCR domains consist of approximately 60 amino acids and are known as a conserved domain in complement regulatory proteins (3). The C-terminal SCR5 of FHR-1 display a high degree of homology (97%) with the C-terminal SCR20 of FH, which contains a surface recognition domain (10, 15–18). As a result, FH and FHR-1 exhibit binding affinity for similar ligands, such as C3b, C3d, and cell surfaces, contributing to their roles in complement system regulation (3, 4, 15–18).
However, significant differences exist between FH SCR20 and FHR-1 SCR5, particularly in their functional implications and regulatory mechanisms. FH SCR20 binds to sialic acid, glycosaminoglycans and C3b, thus driving the recruitment of FH to cell surfaces and to the cell matrix. On the other hand, FHR-1 SCR5 strongly interacts with native C3, attracting native C3 to the proximity of the cell surface. FHR-1 is believed to exert regulatory effects through different pathways, such as modulating FH activity or interacting with other complement components (3, 4, 18, 19).
The ability of FH to bind sialic acid depends on two specific amino acids, S1191 and V1197, located in SCR20 of the FH (4, 17, 18). These residues are unique to FH and are essential for its function (18, 19). In contrast, FHR-1 contains two specific residues, L290 and A296, that are distinct from FH (4, 18, 19). FHR-1 lacks the same regulatory function of FH, which requires the N-terminus (18, 19). The CFH::CFHR1 fusion gene preserves the FHR-1-specific C-terminal residues (18, 19). Consequently, the FH/FHR-1 hybrid protein fails to recruit FH/FHR-1 hybrid protein to cell surface and causes a loss of complement regulation on the cell surface (3, 4, 18, 19).
In addition to the CFH::CFHR1 fusion gene, other fusion genes derived from the CFH–CFHR region have been identified, including CFH::CFHR3 and CFHR1::CFH (3, 8, 11, 12). These genetic variations underscore the complexity of complement regulation and its implications in disorders, such as aHUS.
aHUS develops as a result of vascular endothelial cell damage caused by the activation of the complement cascade; however, not everyone with a CFH::CFHR1 fusion gene develops aHUS. For example, the proband’s father never experienced aHUS, even though the proband’s paternal grandfather developed TMA in his thirties. Interestingly, despite the association of the CFH::CFHR1 fusion gene with a poor clinical prognosis and a high risk of postrenal transplant recurrence, the paternal grandfather never experienced a relapse (8, 11, 12). This suggests that additional factors or triggers are likely necessary to induce aHUS in individuals with the CFH::CFHR1 fusion gene. We have not investigated the aHUS risk haplotypes in CFH and MCP in the patient, father, and grandfather in this study. However, differences in such areas cannot be ruled out as potential contributing factors.
In addition, the patient in this case had a duplication of the CFHR3-1-4-2 genes, which is the first reported case of such a duplication occurring in combination with a CFH::CFHR1 fusion gene. The significance of the CFHR3-1-4-2 gene duplication and increased FHR levels is unclear. One possibility is that FHR competitively inhibit the binding of FH to C3b and glycosaminoglycans, thereby interfering with its normal function (20, 21). Additionally, FHR-1 could also act as a complement-activating molecule (22).
The analysis conducted in the present study revealed three rearranged regions (one in the CFH::CFHR1 fusion gene, one between the CFH::CFHR1 fusion gene and the inserted/original CFHR3, and one between the inserted/original CFHR2 and the original/inserted CFHR3). This indicates that one more residual ambiguous breakpoint can be technically challenging. To address this issue conclusively, it will be necessary to identify more cases with similar genetic variations.
In conclusion, this case highlights the CFH::CFHR1 fusion gene as a potential cause of aHUS with varying severity and age of onset. The presence of a CFHR3-1-4-2 gene duplication as observed in this case is a novel finding that provides insight into the pathogenesis of aHUS.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statementWritten informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsYT: Data curation, Investigation, Writing – original draft, Writing – review & editing. HT: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. TY: Conceptualization, Data curation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. NS: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. SK: Data curation, Writing – original draft, Writing – review & editing. HF: Data curation, Writing – original draft, Writing – review & editing. YH: Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SM: Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. NK: Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. NI: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Supervision, Writing – original draft, Writing – review & editing. TW: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. HT received partial financial support from 2023 Wakayama Medical Award for Young Researchers.
AcknowledgmentsWe thank Drs. Chihiro Taniguchi, Masaki Fukuda, and Raita Araki for clinical management of the patient.
Conflict of interestNI has received speaker honorarium and research funding from Alexion Pharmaceuticals. However, Alexion Pharmaceuticals had no control over the interpretation, writing, or publication of this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1360855/full#supplementary-material
References3. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: State of the art. Mol Immunol. (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Sánchez-Corral P, Pouw RB, López-Trascasa M, Józsi M. Self-damage caused by dysregulation of the complement alternative pathway: relevance of the factor H protein family. Front Immunol. (2018) 12:1607. doi: 10.3389/fimmu.2018.01607
CrossRef Full Text | Google Scholar
5. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2007) 18:2392–400. doi: 10.1681/ASN.2006080811
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Díaz-Guillén MA, Rodríguez de Córdoba S, Heine-Suñer D. A radiation hybrid map of complement factor H and factor H-related genes. Immunogenetics. (1999) 49:549–52. doi: 10.1007/s002510050534
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. (2006) 108:1267–79. doi: 10.1182/blood-2005-10-007252
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PloS Med. (2006) 3:e431. doi: 10.1371/journal.pmed.0030431
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant. (2011) 26:739–41. doi: 10.1093/ndt/gfq658
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Piras R, Valoti E, Alberti M, Bresin E, Mele C, Breno M, et al. CFH and CFHR structural variants in atypical Hemolytic Uremic Syndrome: Prevalence, genomic characterization and impact on outcome. Front Immunol. (2023) 13:1011580. doi: 10.3389/fimmu.2022.1011580
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Francis NJ, McNicholas B, Awan A, Waldron M, Reddan D, Sadlier D, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood. (2012) 119:591–601. doi: 10.1182/blood-2011-03-339903
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Eyler SJ, Meyer NC, Zhang Y, Xiao X, Nester CM, Smith RJ. A novel hybrid CFHR1/CFH gene causes atypical hemolytic uremic syndrome. Pediatr Nephrol. (2013) 28:2221–5. doi: 10.1007/s00467-013-2560-2
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Rodríguez de Córdoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sánchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. (2004) 41:355–67. doi: 10.1016/j.molimm.2004.02.005
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Yoshida Y, Miyata T, Matsumoto M, Shirotani-Ikejima H, Uchida Y, Ohyama Y, et al. A novel quantitative hemolytic assay coupled with restriction fragment length polymorphisms analysis enabled early diagnosis of atypical hemolytic uremic syndrome and identified unique predisposing mutations in Japan. PloS One. (2015) 10:e0124655. doi: 10.1371/journal.pone.0124655
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and c3 glomerulopathy. J Am Soc Nephrol. (2020) 31:241–56. doi: 10.1681/ASN.2019050515
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. (2015) 26:209–19. doi: 10.1681/ASN.2013121339
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Lucientes-Continente L, Márquez-Tirado B, Goicoechea de Jorge E. The Factor H protein family: The switchers of the complement alternative pathway. Immunol Rev. (2023) 313:25–45. doi: 10.1111/imr.13166
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Papp A, Papp K, Uzonyi B, Cserhalmi M, Csincsi ÁI, Szabó Z, et al. Complement factor H-related proteins FHR1 and FHR5 interact with extracellular matrix ligands, reduce factor H regulatory activity and enhance complement activation. Front Immunol. (2022) 13:845953. doi: 10.3389/fimmu.2022.845953
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A. (2013) 110:4685–90. doi: 10.1073/pnas.1219260110
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Márquez-Tirado B, Gutiérrez-Tenorio J, Tortajada A, Continente LL, Caravaca-Fontán F, Malik TH, et al. Factor H-related protein 1 drives disease susceptibility and prognosis in C3 glomerulopathy. J Am Soc Nephrol. (2022) 33:1137–53. doi: 10.1681/ASN.2021101318
留言 (0)