
記住我

Hepatocellular adenoma (HCA) is a paradigmatic entity for morpho-molecular tumor subtyping. It mainly affects (younger) women without a pre-existing liver disease and is associated with exposure to steroid hormones [1]. In addition, metabolic (e.g., obesity, glycogenosis) and vascular liver diseases may induce HCA formation. HCA subtyping has relevance in terms of potential complications as well as clinical management (Table 1).
HNF1A-inactivated HCAs (H-HCAs) are characterized by prominent fatty change and are negative for fatty acid binding protein 1 (FABP1) by immunohistochemistry [2]. While they show no increased risk of malignant transformation in general, a CTNNB1-independent malignant transformation has been described in patients older than 60 years with lesions > 5 cm in diameter [3]. So far, the transformation risk has not been linked to specific HNF1A mutations. Thus, sequencing of the HNF1A gene is currently neither required for diagnosis nor risk assessment regarding malignant transformation. Of note, liver adenomatosis may be observed in patients with HNF1A germline mutations (who may also develop maturity-onset diabetes of the young type 3).
Inflammatory HCA (I-HCA) results from various mutations in genes contributing to activation of IL-6 signaling [1]. It has some peculiar histological features: inflammatory foci, sinusoidal dilatation, and portal tract-like structures harboring ductular proliferations [2]. Positivity of acute phase proteins (e.g., serum amyloid A, C-reactive protein) compared to the surrounding liver tissue can be used as a diagnostic immunomarker. The tumor-associated secretion of acute phase proteins may result in a systemic inflammation, which can be treated by HCA resection [4].
Activating mutations of the CTNNB gene characterize a subgroup of HCA, which carries an increased risk of malignant transformation into HCC (so-called ß-catenin-activated HCA, B-HCA) [1, 5,6,7]. About half of all B-HCA reveal additional features of inflammatory HCA (BI-HCA) [1, 8]. Overall, the frequency of CTNNB1 mutation in HCA is 10 to 15% [1]. Most mutations affecting exon 3 result in high activity of WNT signaling, while mutations in exons 7 (K335) and 8 (N387) and the S45 mutation in exon 3 lead to weaker pathway activation [6]. The combination of glutamine synthetase (GS) and CD34 immunohistochemistry is able to discriminate these mutations in most cases, but molecular testing is advisable. HCA with classical exon 3 mutations show a diffuse GS expression and increased sinusoidal CD34 expression. Exon 3 S45 mutation is characterized by heterogeneous GS staining associated with a GS-positive but CD34-negative rim, while the central lesion reveals a diffuse capillarization. Exon 7/8 mutations show a similar CD34 staining pattern, but GS positivity is only focal and patchy [9]. Strong activation of WNT signaling resulting from classical exon 3 mutations or S45 allele duplication has been associated with a high risk of malignant transformation [6]. Consequently, molecular testing not only clarifies the precise nature of the CTNNB gene mutation but it also provides information about the risk of malignant transformation and is thus predictive in terms of therapeutic decisions (resection of all B-HCA with high WNT-pathway activation).
A rare HCA subtype reveals activation of sonic hedgehog signaling (SH-HCA) due to focal deletions that fuse the promoter of INHBE with GLI1. These tumors occur more frequently in obese patient and have a higher risk of rupture and life-threatening bleeding [1]. Argininosuccinate synthase 1 has been proposed as a diagnostic immunomarker [10]. The very recently described familial adenomatous polyposis (FAP)-HCA occurs in patients with germline mutations of the APC gene and shows also activation of the WNT signaling pathway as demonstrated by strong positivity for glutamine synthetase. Thus, this rare subtype shares features with B-HCA, but it does not reveal nuclear beta-catenin accumulation and an increased risk of malignant transformation has not been established for these HCA [11]. Finally, rare HCAs that do not fit in the above-mentioned subtypes are considered unclassified HCA (U-HCA).
Hepatocellular carcinomaNumerous more or less differentiated attempts to subclassify hepatocellular carcinoma (HCC) using molecular genetic testing, expression profiling (RNA- and protein-based), epigenetics, and combinations thereof have been made. These analyses have uncovered molecular mechanisms contributing to different modes of HCC development and have thus provided the basis for further research. None of these approaches has made its way into HCC diagnosis or clinical management of HCC patients, as they have several shortcomings: there are many proposals but no consensus regarding classification schemes and methodology. As only earlier (resectable) tumor stages have been included, it is unclear whether the classification schemes represent molecular tumor typing or staging and to which extent they are valid for progressed HCCs.
Table 1 Clinico-pathological features of HCA subtypesIt has become apparent that HCC, besides the majority of typical HCCs showing different growth and cytological patterns (may be called HCC, not otherwise specified), contains several specific morpho-molecular subtypes, which show peculiar histological, molecular, clinical, and biological characteristics (Table 2) and whose phenotypes typically remain stable throughout tumor progression. HCC subtyping has been included into the 5th edition of the World Health Organization (WHO) classification. Molecular analyses may support the diagnosis of these subtypes in questionable cases, of, e.g., fibrolamellar, sclerotic, or chromophobe HCCs. Other subtypes, such as lymphocyte-rich and steatotic subtypes, are still less clearly defined and lack consented diagnostically applicable molecular markers.
Table 2 WHO subtypes of HCC [24]In rare cases, the demonstration of hepatitis B virus-DNA integrations in HCC may establish a causal relation between profession-based infection and HCC development.
CholangiocarcinomaIntrahepatic cholangiocarcinoma (iCCA) is the second most frequent malignant primary liver tumor entity and needs to be separated from carcinomas of the gallbladder (GBCA) and extrahepatic biliary tree (eCCA). As suggested by animal models, iCCA may (under certain conditions also) develop from hepatocytes [12, 13]. In line with this hypothesis, detection of albumin mRNA expression by albumin in situ hybridization has been proposed to support the diagnosis of small duct (sd)-iCCA [14, 15].
At the histological level, a sd-iCCA composed of non-mucinous, cuboidal cells forming small tubular and ductular structures in a desmoplastic stroma is separated from a large duct (ld) type, which is biologically similar to eCCA and contains mucin-secreting, columnar cancer cells. sd-iCCA shares the etiological risk profile and primary nodular growth pattern with HCC, while ld-iCCA mirrors the etiology and growth pattern of eCCA.
While eCCA and iCCA share some common mutations (e.g., TP53, BRCA1, BRCA2, PK3CA, KRAS, SMAD4, ARID1A, GNAS), others are especially typical for small duct-iCCA (IDH1, IDH2, and BAP1 mutations as well as translocations involving FGFR2, NRG1, ALK, NTRK1-3, and possibly others; Table 3) and may eventually allow for the identification of iCCA in a cancer of unknown primary (CUP) constellation [16, 17]. This is of direct clinical relevance as a specification of a hepatic adeno-CUP may provide the patient access to several specific guideline- and approval-based targeted and non-targeted therapeutic options superior to standard adeno-CUP chemotherapy.
Table 3 Mutational spectrum of eCCA and iCCA (modified according to [58]There is the first evidence that small duct iCCA, similar to HCC, may contain to a significant extent various different, low-frequency, morpho-molecularly defined subtypes; next to the cholangiolocellular and ductal-plate malformation-like subtypes that are already recognized, the solid-tubulocystic (“cholangioblastic”) subtype with its peculiar morphology, inhibin-positivity, and diagnostic NIPL-NICC1 translocation has recently been defined [18]. Further potential morpho-molecular subtypes have been proposed and await confirmation.
Pancreatic cancerSeveral attempts to molecularly subclassify pancreatic ductal adenocarcinoma (PDAC) have been carried out based on microarray data obtained from cell lines and PDAC tissue, RNA-sequencing in silico subtraction of transcript data obtained from cells comprising the tumor microenviroment (TME). [16, 17, 19]. Based on these results and additional studies [18,19,20], there is now some consensus that acknowledges at least two different molecular subtypes with some overlap and inter- and intra-tumor heterogeneities (classic and basal types). Several clinical studies aim at harnessing molecular subtypes as well as other RNA-based signatures to inform efficacy of systemic treatments. Alternatively, assessment of copy number variations (CNVs) and larger chromosomal rearrangements can classify PDAC into four subtypes: “stable,” “locally rearranged,” “scattered,” and “unstable” [20]. These may be exploited clinically in the future, as the unstable subtype is associated with homologous recombination deficiency and there is evidence that CNV-rich tumors tend to display a cold TME. Currently, molecular testing is not required clinically to define or subtype PDAC.
Other tumor entitiesIn other liver tumor entities, molecular testing is rarely required for typing. Rare exceptions may be questionable cases of malignant epithelioid hemangioendothelioma. Here, the demonstration of a WWTR1-CAMTA1 fusion gene (up to 90% of the cases) or the rare YAP1-TFE3 translocation may support the diagnosis in questionable cases [21]. In cases of hepatic adeno-CUP, the pattern of molecular alterations may provide valuable information in regard to the entity. For example, detection of an FGFR2 translocation characterizes the tumor as a sd-iCCA with high certainty and provides the patient with several approved systemic therapeutic options.
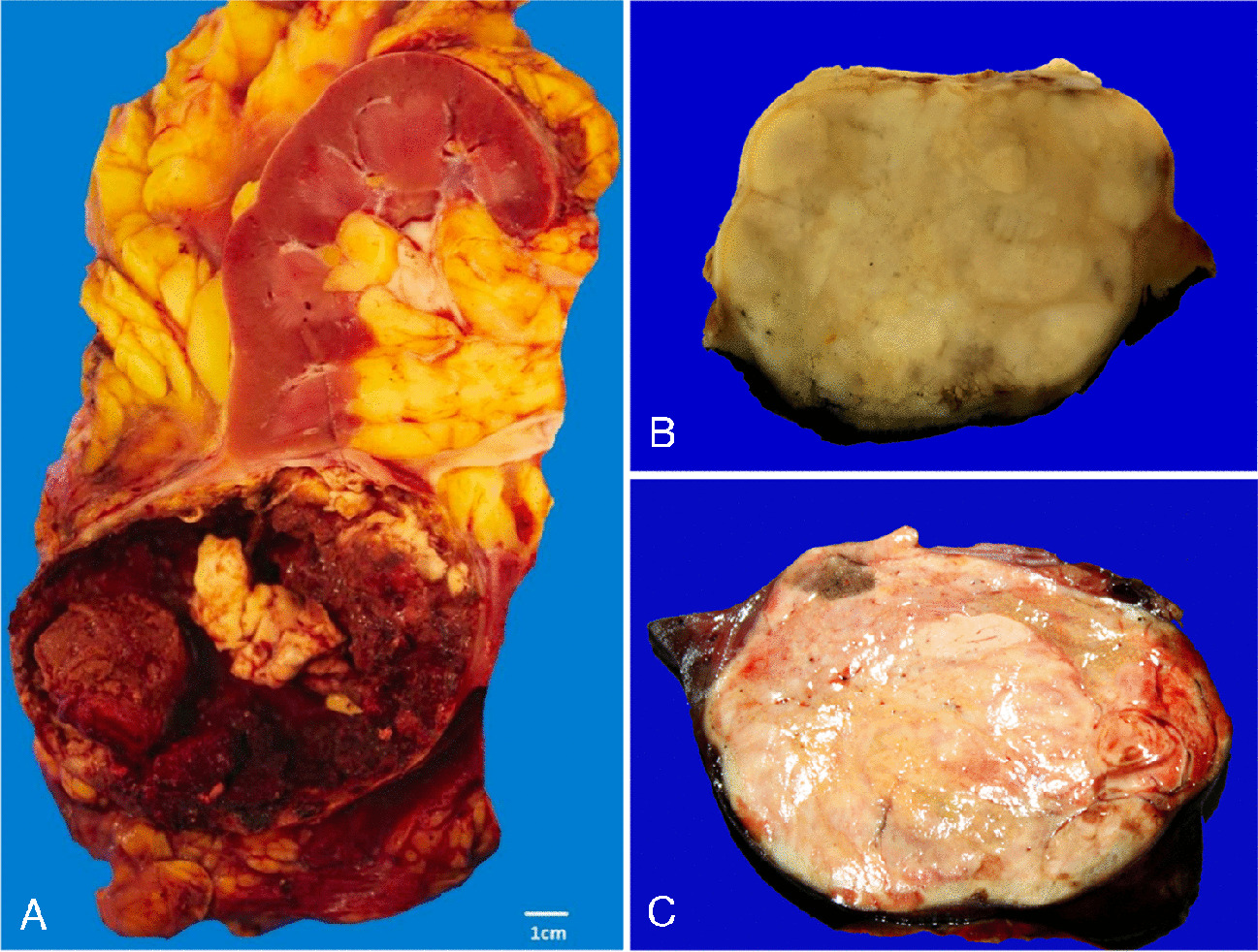
Definition of malignancyTumor typing includes the reliable distinction of benign or premalignant hepatocellular lesions from early and highly differentiated HCC. One scenario is malignant transformation of B-HCA into HCC and the other the differential diagnosis between premalignant dysplastic nodules and highly differentiated HCC (Fig. 1).
Fig. 1
Modes of malignant transformation of HCA are shown in the upper panel. While HCC development on the basis of B-HCA and BI-HCA relies on the presence of the CTNNB1 gene mutation and further acquired alterations, malignant transformation of (mostly HNF1A inactivated) HCA in older patients occurs in setting of a wild-type CTNNB1 gene locus. The development of high-grade dysplastic nodules in cirrhotic livers is shown in the lower panel and is associated with chromosomal gains at 1q or 8q. Frequent mutations involving the transition from dysplastic nodule to HCC are shown in black, while altered candidates potentially useful as drug targets are shown in gray. Abbreviations: B-HCA, beta-catenin-activated HCA; BI-HCA, beta-catenin-activated inflammatory HCA; chr, chromosome; CTNNB1, beta-catenin gene; EGFR, epidermal growth factor receptor; FGF19, fibroblast growth factor 19; HCA, hepatocellular adenoma; HNF1A, hepatocyte nuclear factor 1A; GLI1, GLI family zinc finger 1; IL-6, interleukin-6; INHBE, inhibin subunit beta E; MET, MET proto-oncogene; MSI-H, high microsatellite instability; PI3K, phosphoinositide-3-kinase; RB1, RB transcriptional corepressor 1; TERT, telomerase reverse transcriptase; TP53, tumor protein P53; VEGFA, vascular endothelial growth factor A
Of note, the histological changes in the surrounding liver tissue as caused by the underlying chronic liver disease together with the so-called matrix diagnosis (e.g., older age, male gender, presence of chronic liver disease, patient origin from high-risk area) may support the diagnosis of HCC vs. HCA or FNH. Since definite histopathological features of malignant transformation (interstitial and vascular invasions) are rarely found in a critical biopsy specimen, next to the demonstration of disturbed trabecular architecture, the diagnosis of well-differentiated HCC may be supported by diffuse capillarization of the sinusoids in HCC as detected by CD34 immunohistology [22]. In addition, an immunohistological marker panel (heat shock protein 70, glypican-3, and GS; Table 4) became the diagnostic standard for the molecular adjunct diagnosis for malignancy in highly differentiated hepatocellular tumors. It provides a high sensitivity (~ 70%) and a near perfect specificity for the detection of malignancy in independent studies [23]. Moreover, detection of mutations in the telomerase reverse transcriptase promoter may be helpful for the identification of malignant transformation in highly differentiated hepatocellular tumors, namely, HCA and dysplastic nodules vs HCC [7]. It employs the fact that hTERT promoter mutations significantly increase in frequency from HCA (0%) to “borderline” cases (17%) to HCC derived from HCA (56%) [7] and from dysplastic nodules (6–19%) to early and highly differentiated HCC (43–61%) [24]. It has to be noted that the detection of hTERT-promoter mutations requires specific DNA-PCR-based assays and cannot be detected by standard panel-based assays or WES.
Table 4 Diagnostic performance of HCC marker panel (modified according to [59]In about 85–90% of the cases, PDAC may develop from different premalignant precursors: pancreatic intraepithelial neoplasia (PanIN) progresses from low- to high-grade lesions accumulating genetic alterations (e.g., mutations in KRAS, SMAD4, TP53) [25, 26]. Furthermore, intraductal papillary mucinous neoplasms (IPMNs) may progress from low-grade to high-grade dysplasia to PDAC, and more rarely mucinous cystic neoplasms (MCNs) may malignantly transform. KRAS mutations can be observed early in neoplastic development (i.e., even in low-grade PanIN and IPMN) consequently increasing to more than 90% of PDAC carrying activating KRAS mutations as major driver event. While diagnosis of malignant transformation in pancreatic carcinogenesis resides solely on histology not requiring any molecular testing, molecular testing of aspiration fluid may help to clarify the nature of cystic pancreatic lesions [27, 28].
Genetic cancer syndromes and genetic tumor predispositionAffection of the liver and biliary tree in genetic cancer syndromes is very rare. The vast majority of cases of HCC with a genetic background are due to hereditary metabolic diseases (e.g., genetic iron storage diseases, hereditary tyrosinemia type I (rare), Wilson’s disease (rare)). In these cases, severe hepatic disease manifestation provides the soil for HCC and iCCA development. Consequently, prevention of liver affection abolishes the risk of tumor development. The reason for the relative protection in regard to hepatic involvement in genetic tumor predisposition syndromes is unknown. Thus, there is no indication to test for genetic cancer syndromes in HCC and CCA. Genetic predisposition by respective germline mutations is likely in hepatic adenomatosis (> 10 HCA in a patient) or when histology or immunohistology detect multiple comparable microlesions in the non-tumorous parenchyma of a HCA-resection specimen. Angiomyolipoma (AML) has been linked to the tuberous sclerosis complex, but this correlation is much lower in hepatic AML when compared to renal AML. Rarely, hereditary cases of pancreatic cancer have been observed in association with Peutz-Jeghers syndrome (STK11), hereditary pancreatitis (PRSS1, SPINK1, CFTR), familial melanoma (CDKN2A, CDK4, BAP1), Lynch syndrome (MLH1, MSH2, MSH6, PMS2), hereditary breast and ovarian cancer syndrome (BRCA2, BRCA1, PALB2), Li-Fraumeni syndrome (TP53), FAP (APC), ataxia telangiectasia, and polymerase proofreading-associated polyposis (POLE, POLD1) [29, 30].
留言 (0)