
記住我
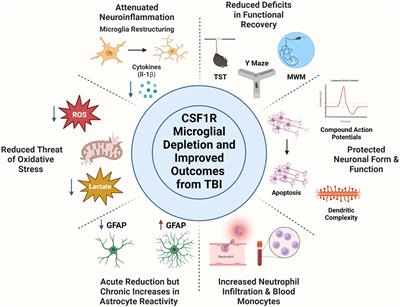



Traumatic brain injury (TBI) is one of the most common types of brain injury, with major medical and socio-economic problems (Hyder et al., 2007; Rubiano et al., 2015). It is approximated that TBI annually afflicts a range of 54 to 60 million individuals, necessitating hospitalization or culminating in mortality. Worldwide, TBI accounted for 27.16 million new cases (with 95% uncertainty intervals of 23.36 to 31.42 million), 48.99 million prevalent cases (46.84 to 51.32 million), and 7.08 (5.00 to 9.59 million) million years lived with disability (YLDs) in 2019 (Guan et al., 2023). On a global scale, the age-standardized incidence rates of TBI have demonstrated a significant decrease since 1990. This positive trend suggests potential benefits arising from international initiatives aimed at lowering TBI rates. Notably, substantial efforts have been directed toward enhancing road safety and mitigating road-related injuries, a prominent contributor to TBI globally. Among the diverse injury categories, cerebral injuries exhibit a heightened propensity to resulting in either death or sustained disabilities (Hyder et al., 2007). The primary factors contributing to TBI vary by age, socio-economic conditions and geographical locations. Low and medium-income countries report an approximately threefold higher proportional incidence of TBI compared to high-income countries. The Southeast Asian and Western Pacific regions bear the highest overall burden in this regard (Dewan et al., 2018). According to studies, falls have been identified as the predominant etiological factor for TBI (74%), followed by road injuries (19%, mainly Africa and Southeast Asia), violence (2%, South America, Caribbean and Sub Saharan Africa), and sports- and work-related incidents (Iaccarino et al., 2018). Over 25% of individuals with long-term mild traumatic brain injury (mTBI) consequences struggle to return to work even a year post-injury (Langlois et al., 2006). Chronic mTBI represents a formidable health challenge characterized by lifelong disabilities and enduring consequences, significantly diminishing the affected individuals’ quality of life (Seabury et al., 2018). The economic ramifications are noteworthy; the estimated overall healthcare cost attributable to non-fatal TBI in 2016 was, with rehabilitation costs per patient reaching $36.000, contributing to an annual national expenditure of nearly $40.6 billion (Miller et al., 2021). Despite these profound implications, the molecular mechanisms underpinning chronic mTBI symptoms remain elusive within current scientific understanding.
Traumatic brain injury is a complex process, in which the primary injury followed by the secondary injury evokes pathological structural changes combined with functional deficits (Galgano et al., 2017). The term primary injury refers to an initial insult caused by mechanical forces, that depending on the intensity/severity and type of injury, may result in contusion or penetration of the brain and/or blood-brain barrier (BBB) disruption. The primary injury can evoke a focal or diffuse type of tissue damage. Diffuse injury occurs with a higher frequency than a focal one, however, a combination of both types is common in patients (Skandsen et al., 2010). The secondary tissue damage occurs over a period of time, from minutes up to several days after the primary injury, and is caused by cellular reactions, activation of metabolic pathways associated with neuroinflammation, edema, hypoxia, or tissue atrophy (Ng and Lee, 2019). The damage and the extent of the injury caused by TBI are determined by the severity of the injury, which can be assessed by various methods, including the Glasgow Coma Score (GCS). The GCS scale evaluates three parameters (eye, verbal, and motor responses) and based on the scores, the injury is classified as mild (GCS 13–15), moderate (GCS 9–12), or severe (GCS 3–8) (Jain and Iverson, 2022). The majority of reported TBI in patients is of mild level (Leo and McCrea, 2016), but numbers of cases may be higher as numerous patients with suspected mTBI do not seek medical help after an accident. Additionally, the period following TBI can be divided into three phases: acute (24 h post-TBI), subacute (1 day–3 weeks post-TBI), and chronic phase (more than 3 weeks post-TBI). However, these time periods are only general and may vary from case to case as they are affected by various factors, such as age, injury type and location (Lund et al., 2016; Toshkezi et al., 2018).
Secondary brain injury comprises a complex series of cascading events within the brain. These events include the release of pro-inflammatory cytokines, chemokines, and signaling molecules, which create a pro-inflammatory microenvironment. This environment exacerbates neuronal excitotoxicity and oxidative stress, contributing significantly to the development of secondary brain damage (Ng and Lee, 2019). Glial cells, representing the most abundant brain cells, play a prominent role in these events, as well as in synaptic pruning, synaptic plasticity alterations, and the long-term functional outcomes of the injured brain, emphasizing their central position in the pathophysiology of secondary brain injury (Mira et al., 2021).
In response to tissue trauma, glia cells undergo both morphological and functional changes characterized by the term reactive gliosis, which usually refers to astrocytes and microglia (Buffo et al., 2008; Gao et al., 2013; Pekny and Pekna, 2016). Reactive glia, as part of the lesion, are involved in neuroinflammation and edema to varying degrees, create the glial scar and produce extracellular matrix (ECM) molecules, such as chondroitin sulfate proteoglycans (CSPGs) that contribute to glial scar formation and stabilize it (Asher et al., 2001; Dyck and Karimi-Abdolrezaee, 2015). Moreover, reactive glia produce a large spectrum of proteins, including growth factors and cytokines that are crucial for intercellular cooperation as well as for protective mechanisms and regeneration of damaged tissue (Ziebell and Morganti-Kossmann, 2010).
In the healthy brain, astrocytes are the main effector cells responsible for the optimal environment for neuronal survival and functions: they maintain ion/pH/water homeostasis and BBB integrity, modulate synaptic activity, ensure the neurotransmitter clearance from the extracellular space and are involved in neuronal metabolism, by providing energy substrates as well as by metabolite removal (Kim et al., 2019). During TBI, besides being a key component forming glial scar, reactive astrocytes produce pro-inflammatory cytokines and chemokines (Gyoneva and Ransohoff, 2015; Michinaga and Koyama, 2021). Changes in the expression and localization of various astrocytic proteins forming ion channels and transporters, such as aquaporin-4 (AQP4) or glutamate transporters EAAT1/GLT-1 and EAAT2/GLAST, also contribute to edema development and excitotoxicity (van Landeghem et al., 2006; Beschorner et al., 2007; Kapoor et al., 2013). Unlike astrocytes, the functions of microglia are much more narrowly focused and their main function under physiological conditions is immune surveillance and the phagocytosis of apoptotic debris. However, it has also been demonstrated that microglia can be involved in synapse formation and maintaining synaptic homeostasis, as well as in the production of a variety of growth factors modulating neuronal activity (Borst et al., 2021). In the injured brain, microglia are the key components of neuroinflammation, and together with astrocytes they contribute to scar formation (Wake et al., 2009; Miyamoto et al., 2013). Oligodendrocytes are myelinating cells providing support and insulating myelin sheaths to axons. Pathologies such as TBI accompanied by oxidative stress or excitotoxicity have a detrimental effect on oligodendrocytes, resulting in their apoptosis and demyelination, which can significantly affect the resulting post-TBI functional outcome (Dent et al., 2015). There is increasing evidence of the important role of oligodendrocyte progenitor cells (OPCs), also known as polydendrocytes or NG2-glia, in brain pathologies, including TBI (Dimou and Gallo, 2015; Akay et al., 2021). NG2-glia have a large capacity to proliferate and differentiate into other cellular types, mostly in myelinating oligodendrocytes under physiological conditions (Zhu et al., 2008; Dimou and Gallo, 2015). In the injured brain, NG2-glia become part of the lesion and glial scar, contribute to wound closure, regulate neuroinflammation, and besides oligodendrocytes, NG2-glia can differentiate into other cell types, especially reactive astrocytes which seem to be injury type-dependent (Honsa et al., 2016; Hackett et al., 2018).
Traumatic brain injury research articles are mostly aimed at astrocytes and microglia, while NG2-glia and oligodendrocytes are usually neglected. In this review, we focus on the roles of these four types of glial cells in secondary injury pathologies based on observations in the different TBI models and factors that may impact the outcomes and should be considered when designing experiments.
2 Experimental models of traumatic brain injuryTraumatic brain injury is a highly variable and complex process, and there is currently no animal model which would be able to completely reproduce all the changes that occur after TBI. To date, a number of TBI models have been developed that differ in the type of the induced injury (focal vs. diffuse; impacted vs. non-impacted) as well as in the severity of the tissue damage. Here, we list a brief overview of the most known models: Weight drop injury (WD), Controlled cortical impact (CCI), Fluid percussion injury (FPI), Blast injury (BI), Penetrating ballistic-like brain injury (PBBI) and Close-head impact model of engineered rotational acceleration (CHIMERA) [for comprehensive reviews see (Xiong et al., 2013; McDonald et al., 2021)]. With regards to the necessary equipment, the WD model is the simplest to perform, while the others require sophisticated devices. Rodents are the most used animals for TBI experiments, but animals such as zebrafish (Hentig et al., 2021) and Drosophila (Katzenberger et al., 2013) are used as well. The CCI and WD models can be produced as an open-head or closed-head injury (CHI), FPI and PBBI are open-head type of models, while BI and CHIMERA are CHI models. Open head models require a craniotomy, and the impact is directed to the surface of the dura. CHI is a type of TBI, where the force impacts on the intact skull (Deshetty and Periyasamy, 2023).
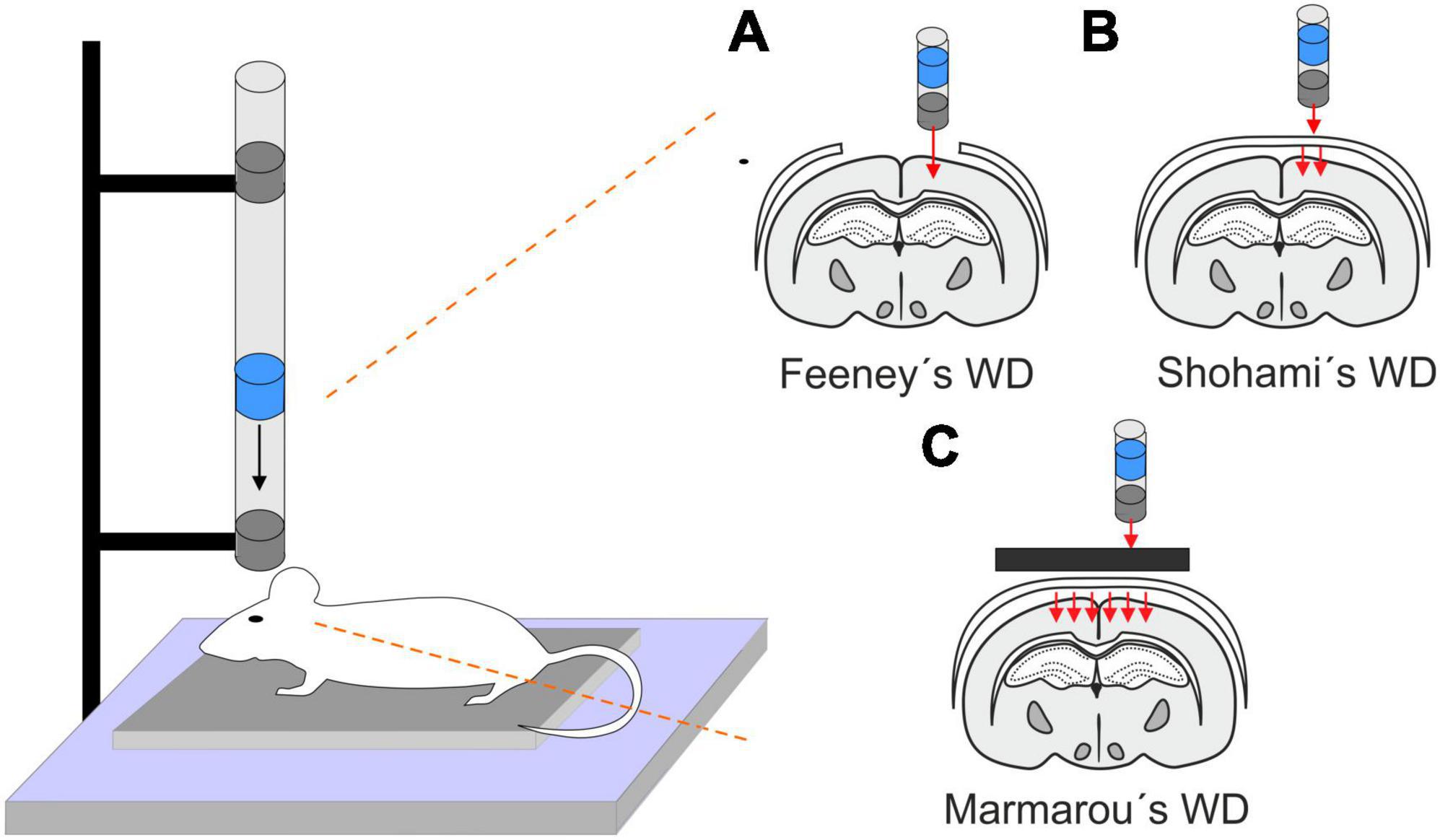
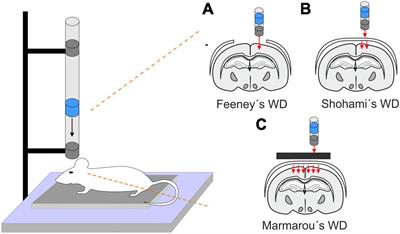
2.1 Weight drop (WD)This model is performed by a free fall of guided weight to an exposed skull (depending on the model, with or without craniotomy) (Figure 1). There are 3 basic types of this model: 1. Feeney’s model exposes the dura, and the weight impact results in cortical contusion followed by the development of a necrotic cavity (Feeney et al., 1981). 2. Shohami’s model is a CHI model, where weight impacts an unprotected skull. Due to this model, edema occurs in the traumatized hemisphere with the addition of hemorrhagic lesions which in turn develop into necrosis (Shapira et al., 1988). 3. The Marmarou’s model uses a metal disc to prevent skull fractions. This model mimics TBI induced by falls and motor vehicle accidents and evokes a diffuse injury without lesion development (Marmarou et al., 1994). The severity of the WD model can be adjusted by changes of the weight mass and/or height of the drop of the weight (Ma et al., 2019). The WD model replicates human TBI fairly well, and the WD device is simple and affordable in comparison to other models. However, complications with skull fractures, high mortality, and variability in observed injuries may emerge (Zhang et al., 2014).
Figure 1. Models of traumatic brain injury - Weight drop (WD). WD models use free fall of guided weight in different modifications. (A) In Feeney’s WD model, the weight drops on the exposed dura. (B) In Shohami’s WD model, the weight impacts an unprotected scull. (C) In Marmarou’s WD model, the weight is dropped on a metal disc or other scull protection and simulates a diffuse concussive head injury for example in football players or car accidents.
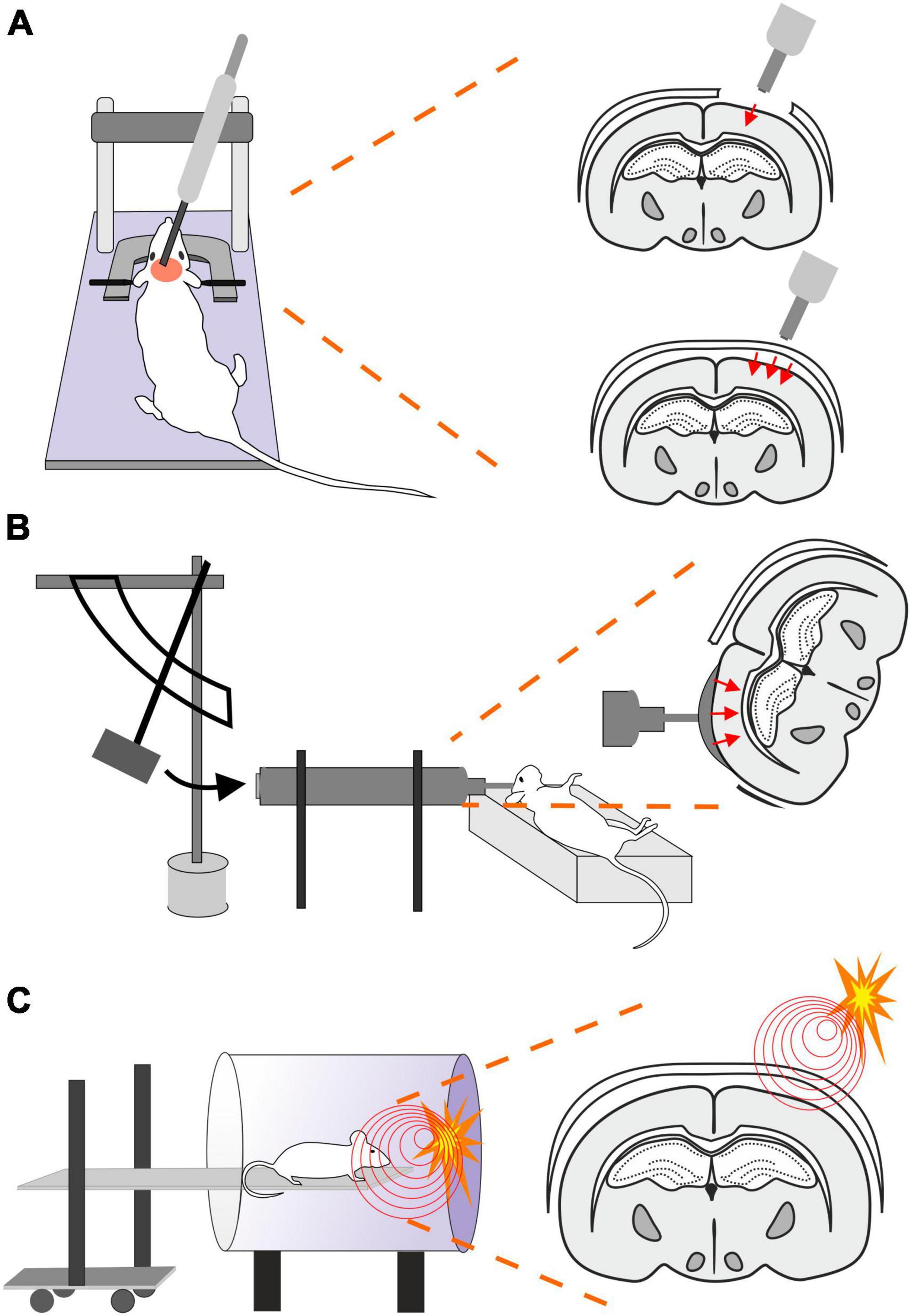
2.2 Controlled cortical impact (CCI)This model uses a controlled piston (electromagnetic or pneumatic control) which delivers an impact to the exposed dura (Osier and Dixon, 2016; Figure 2A). This type of model evokes a focal brain injury resulting in cortical tissue loss, axonal injury, BBB disruption and development of edema. Moreover, this model was adapted to be used for close head injury, CCI-CHI (Romine et al., 2014; Osier and Dixon, 2016). The severity of the resulting injury can be adjusted by controlling the depth impact and the velocity of the piston (Ma et al., 2019).
Figure 2. Models of traumatic brain injury–Controlled cortical impact (CCI), Fluid percussion injury (FPI) and Blast injury (BI). (A) In CCI model, pneumatic- or an electromagnetic-controlled piston impact the exposed dura. The severity of the evoked focal TBI depends on the depth of the impact and the velocity of the piston impacting either the exposed dura (frequently used as a model of focal/open head injury; upper scheme) or unprotected skull (modified CCI to mimick diffuse/closed head injury; lower scheme). Moreover, the TBI severity is also affected by the material of the pad (foam vs. solid) and impactor tip (rubber vs. solid) or by the presence or absence of head fixation. (B) Brain injury in FPI model is evoked by a fluid pulse transferred to exposed dura by the impact of a pendulum on a piston filled with fluid. This mode mimics brain contusion without scull fraction and its outcome depends on the localization of the craniotomy and the angle of the pendulum. (C) In the BI model, the animal is placed on a movable platform in a shock tube capable of controlling overpressure waves evoked by a controlled detonation. The model simulates diffuse mTBI often seen in military personnel.
The advantages of this model are the low mortality rate, no risk of rebound injury, and result reproducibility. However, it also depends on the impactor diameter and whether the simple CCI or adapted CCI-CHI is used (Zhang et al., 2014; Ma et al., 2019).
2.3 Fluid percussion injury (FPI)The FPI model uses a fluid pulse delivered to the dura by the impact of a pendulum on a piston filled with fluid (Ma et al., 2019; Figure 2B). Based on the placement, this technique can be used for lateral FPI (a combination of focal and diffuse injury) or midline FPI (diffuse injury). FPI simulates brain contusion without skull fracture. Localization of the craniotomy is an important factor when using this model. Floyd et al. (2002) tested 4 groups with rostral, caudal, medial, and lateral craniotomy. The caudal group exhibited a higher mortality rate than other groups after FPI, and the rostral group showed the lowest level of cellular loss in the hippocampal area. The authors also showed differences in the levels of reactive astrogliosis in a brain area-dependent manner, whereas the rostral group showed less immunoreactivity for astrocytic marker glial fibrillary acidic protein (GFAP) than the rest of the groups. However, it should be noted that fewer animals per group were used for the GFAP expression tests than for the rest of the aforementioned examinations (Floyd et al., 2002). The FPI model fairly accurately mimics the glial reaction to TBI, as the studies using this model reported hypertrophy of astrocytes with different orientation, and a decreased number of their processes or structural changes of microglia (rod-like morphology) (Ziebell et al., 2012; Robinson et al., 2016). The FPI model can be adjusted by setting the angle of the pendulum higher or lower (Eakin et al., 2015). This model results in direct brain trauma without the necessity of skull protection, and the mortality rate is relatively low. The disadvantage of this model is the necessity of fluid percussion system monitoring during the procedure, since factors such as residual air in the system can cause variability or added injuries (Zhang et al., 2014).
2.4 Blast injury (BI)Blast-induced traumatic brain injury (bTBI) is caused by exposure to explosive devices. This type of injury is often called “invisible injury” because there is no evidence of an external injury (Lucci, 2006). This form of neurotrauma represents the most prevalent type of mTBI in the context of military personnel. Blast waves cause significant oscillating acceleration-deceleration cycles on the head, which is known as the “bobblehead effect” (Rosenfeld et al., 2013). The detonation of an explosive device generates a supersonic pressure gradient, caused by a primary blast wave. This wave consists of a high positive pressure followed by a prolonged negative pressure phase, forming a Friedlander curve. Primary bTBI is characterized by internal injuries that are challenging to detect and assess for severity. Strong blasts can cause acute injury or death, while exposure to milder forces may result in delayed or subclinical neuropathological changes. To simulate bTBI, a shock tube capable of controlling overpressure waves is utilized to mimick real-life free-field explosions, resembling the Friedlander-type waveform (Huber et al., 2013; Campos-Pires et al., 2018; Figure 2C).
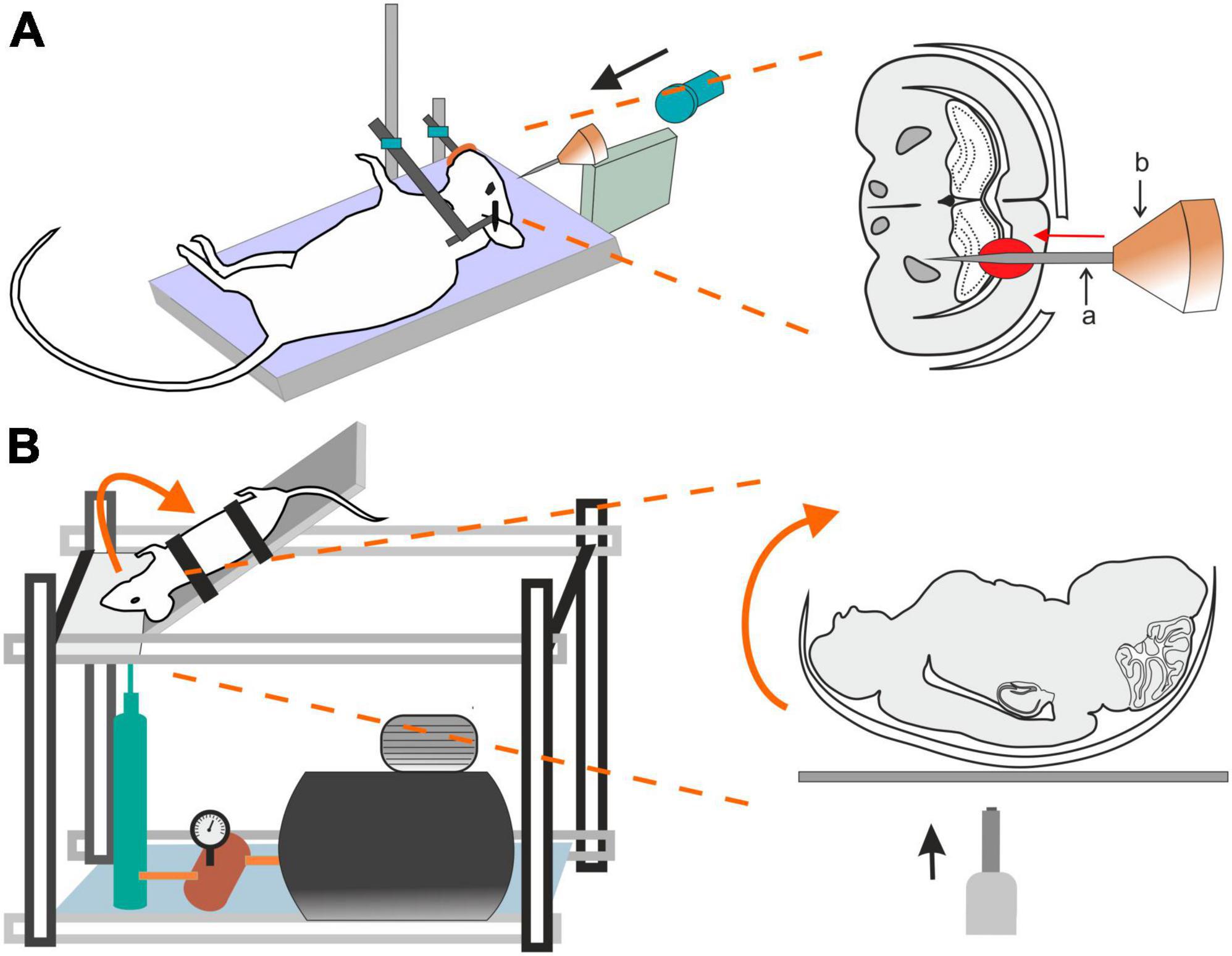
2.5 Penetrating ballistic-like brain injury (PBBI)Penetrating ballistic-like brain injury (PBBI) results from high-energy projectiles, creating a substantial temporary cavity in the brain (Williams et al., 2005, 2006; Figure 3A). The outcome is closely linked to the projectile’s anatomical path and energy transfer (Williams et al., 2005). Using this model in rats results in cognitive impairment, white and gray matter damage, brain swelling, seizures, cortical spreading depression, and neuroinflammation (Williams et al., 2007). Studies using new rodent PBBI models, including a non-fatal model which uses a modified air rifle for low-velocity PBBI or another PBBI model simulating the effect of high-energy projectiles, reveal immediate and subacute changes such as increased intracranial pressure, BBB permeability, brain edema formation, and persistent motor and cognitive deficits (Plantman et al., 2012). The unique aspect of extensive intracerebral hemorrhage makes PBBI a valuable model for studying moderate-to-severe brain trauma as well as for assessing the effect of therapeutic interventions (Williams et al., 2006; Chen et al., 2009; Shear et al., 2009).
Figure 3. Models of traumatic brain injury–Penetrating ballistic-like brain injury (PBBI) and the Closed-head impact model of engineered rotational acceleration (CHIMERA). (A) In the PBBI model, a modified air-rifle is utilized to accelerate a projectile (led pellet) into a conic impactor probe (a) placed in an impactor holder (b) against the animal head which is fixed by stereotactic apparatus and a nose clip. Probe penetration results in the creation of temporary brain cavity, mimicking the damage after a gun shot. The severity of the injury can be modified by adjusting the air-rifle pressure. (B) In the device utilized for a mouse CHIMERA, a piston, driven by a high-pressure pulse, impacts the plate which the animal’s head is freely resting on, while the animal body is fixed in a supine position. The impact results in a sagittal plane motion of the animal head.
2.6 Closed-head impact model of engineered rotational acceleration (CHIMERA)This model is highlighted for its relevance in studying the impact of the acceleration effects associated with major trauma, including falls, vehicular accidents, and sports-related collisions (Namjoshi et al., 2014) and is particularly important for modeling axonal and white matter injury (Bashir et al., 2020; Krieg et al., 2023). CHIMERA’s commercial availability is emphasized, providing researchers with the means to standardize injury parameters and ensure consistent replication across various laboratories. Specifically, the mouse-oriented CHIMERA device exhibits compressive contact and inertial forces, along with changes in velocity and angular velocity akin to values observed in professional football and boxing (Namjoshi et al., 2014).
The CHIMERA TBI model utilizes the high-pressure impact from a metal piston on the platform under the animal’s head (Figure 3B). While the animal head rests freely, its body is secured in a supine position with straps, aided by crosshairs for precise head alignment. Compressed air, regulated by a valve, propels the piston, and its velocity is measured by infrared devices. A pressure gauge regulates impact intensity. Upon device activation, the ascending platform initiates a forward swing of the animal head that touched the sternum before returning to the original position on the platform (McNamara et al., 2020). All CHIMERA models involve animals in a supine position, causing sagittal plane motion. CHIMERA device reconfiguration enables a comparison of sagittal and horizontal impacts, shedding light on varying functional and morphological outcomes associated with different injury planes (Browne et al., 2011; Mychasiuk et al., 2016).
2.7 Repeated mild traumatic brain injuryThe protocols of WD, CCI, FPI, and BI can be adapted to produce only mTBI by adjustment of the velocity, depth of duration of a traumatic pulse/impact. To simulate the head trauma often seen in contact sports, models of repetitive mild traumatic brain injuries (rmTBIs) have been introduced, where each single injury is below the concussion threshold, but their effects add up and may result in prolonged and/or delayed tissue damage and neurological deficit. The parameters of experimental protocols and observed alteration in biochemical/neuropathological analysis as well as in neurological outcome in different studies is thoroughly described in a systematic review of Hiskens et al. (2019).
2.8 Pediatric traumatic brain injury modelsTraumatic brain injury is a significant global issue in infants and children. Head injuries in children can lead to a variety of traumatic injuries to the scalp, skull, and brain, mirroring those in adults. The necessity to study developmental TBI and its long-term consequences in appropriate models arose from significant distinctions in blood flow, metabolic rate, neurotransmitter activity, degree of myelination, ability to tolerate oxidative stress, and/or biomechanical scull properties between adult and the pediatric population (Margulies and Thibault, 2000; Morrison et al., 2013; Araki et al., 2017). For simulation of pediatric TBI, already established modified models such as WD, FPI, CCI, CCI-CHI, CHIMERA, and rmTBI models are employed as well as specific models for abusive head trauma (AHT), commonly known as shaken baby syndrome, which is the predominant form of TBI in infants <1 year (Bonnier et al., 2004). Experimental animals include rodents aged 11–21 days, covering responses from a term infant (aged 7–11 days) to a toddler (aged 17–21 days), piglets (especially in FPI and CHIMERA models) and lambs (shake injury models). Further insights into this topic are provided by comprehensive reviews focusing on the considerable challenges in the field and highlighting essential models that investigate the unique injury mechanisms related to pediatric TBI (Araki et al., 2017; Kochanek et al., 2017; Nwafor et al., 2022).
3 The response of glial cells to traumatic brain injuryBoth human brain trauma as well as its experimental models induce a typical response of nervous tissue that is based predominantly on glia reaction and include:
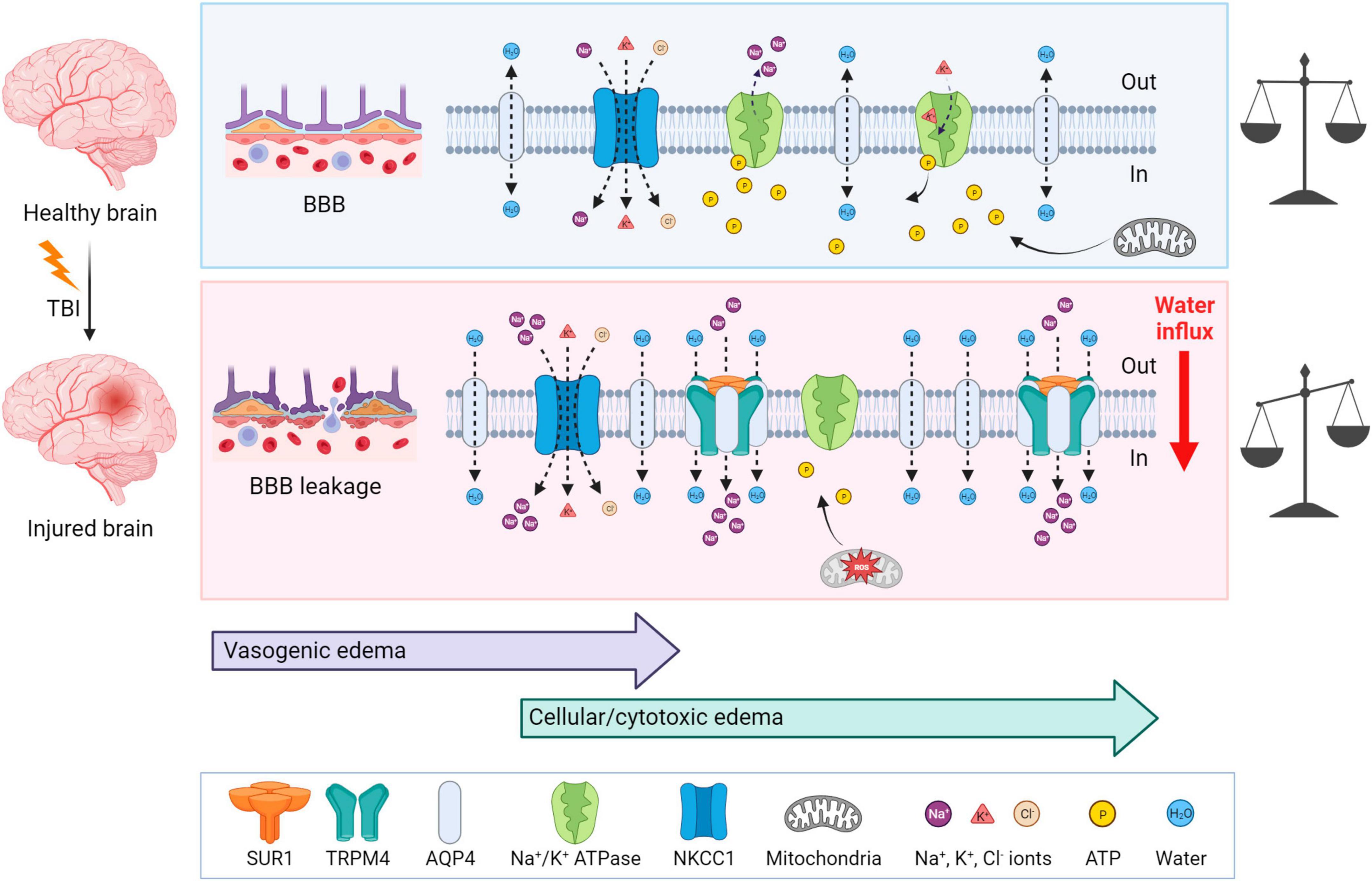
3.1 EdemaTraumatic brain injury is typically accompanied by cerebral edema, which is characterized by an increase and retention of CNS water content, contributing to elevated intracranial pressure. Two types of edema can be present post-TBI: vasogenic and cellular/cytotoxic edema. The vasogenic edema accompanies the compromised BBB integrity after TBI and it is characterized by an extracellular accumulation of fluid. The vasogenic type of edema prevails in the first days post-TBI; while the disrupted BBB gradually closes, cytotoxic edema with a slower onset follows (Donkin and Vink, 2010; Figure 4). Cytotoxic edema, the most common type of edema found in TBI patients (Marmarou et al., 2006), results in cellular swelling (including astrocytes), which can eventually result in apoptosis. However, this type of edema does not contribute to brain swelling or an increase in intracranial pressure.
Figure 4. Mechanisms of brain edema development after traumatic insult—a comparison with a healthy brain. Upper panel: In physiological conditions, ionic balance is ensured by active or passive transport via channels, and mitochondria produce enough ATP to secure normal cellular function. Lower panel: At first, TBI induces BBB disruption and vasogenic edema, which is followed by cytotoxic edema with a slower onset. After TBI, AQP4, SUR1, and TRPM4 are upregulated and create a complex which contributes to astrocytic swelling by the intracellular transport of Na+ and H2O. NKCC1 during TBI is more activated and transports additional ions into the cells, particularly Na+. Moreover, due to the mitochondrial dysfunction induced by ROS, ATP is depleted. Lack of ATP inhibits the active transport via Na+/K+ ATPase and also terminates the ATP-dependent inhibition of SUR1-TRMP4 complex that transport Na+ ions. These events cause water influx and energetic and ionic disbalance, which can consequently result in cell death. TBI, traumatic brain injury; BBB, blood-brain barrier; NKCC1, Na+, K+, Cl– cotransporter; ROS, reactive oxygen species; SUR1, sulfonylurea receptor 1; TRPM4, transient receptor potential melastin 4. Created with BioRender.com.
Cytotoxic edema is characterized by cellular swelling, which arises from an intracellular influx of ions and water. The astrocytes express various channels contributing to water and ion homeostasis, such as aquaporins, NKCC1 (Na+, K+, Cl– cotransporter), Na+/K+ ATPase, or SUR1-TRPM4 (Sulfonylurea receptor 1–transient receptor potential melastin 4) channels (Figure 4). ATP-dependent ion pump functionality is tightly connected with ATP availability. During TBI, generated reactive oxygen species (ROS) cause mitochondrial dysfunction leading to impairment in ATP synthesis and thus to ATP depletion. Moreover, ATP is released from cells into the extracellular space immediately following TBI, therefore exacerbating the energetic disbalance (Moro et al., 2021). The authors also demonstrated that this ATP release can be slowed down significantly by blocking P2Y1 receptors or store-operated calcium channels (Moro et al., 2021). Hence, the ATP-dependent Na+/K+ pump, creating membrane potential by maintaining the Na+/K+ difference across the membrane, fails in providing transport of the ions, causing a change in the membrane potential, followed by an increase in osmolarity and water influx into the cells (Zusman et al., 2020).
Aquaporin-4 (AQP4), a channel participating in water homeostasis and transport, is also considered to be an important contributor to cytotoxic edema. AQP4 is predominantly expressed by astrocytes and enriched in their endfeet in contact with blood vessels (Neely et al., 2001; Amiry-Moghaddam et al., 2004). TBI causes the upregulation of AQP4 expression, which consequently induces the swelling of astrocytes (Kapoor et al., 2013). The degree of edema is dependent on the subcellular localization of AQP4; translocation of AQP4 mediated by calmodulin leads to increased water flux and astrocytic swelling (Kitchen et al., 2020). In AQP4 knockout mice, the migration of astrocytes to the injury site was reduced (Saadoun et al., 2005). Additionally, in wild-type astrocytes, AQP4 is polarized to the leading edge of the membrane and astrocytic migration can be enhanced by the extracellular osmotic gradient. Therefore, authors suggested, that AQP4 is involved in astrocytic cell migration toward the damaged areas after TBI (Saadoun et al., 2005).
Other contributors to astrocytic swelling include the increased activation of NF-κB (Nuclear factor κB) (Jayakumar et al., 2014) and the increased activation of NKCC1 cotransporter in astrocytes, which controls the transport of Na+, K+ and Cl– (Jayakumar et al., 2011). SUR1 is an ATP-binding part of various ion channels, serving as their regulatory subunit. SUR1 is associated with pore-forming subunits, such as Kir6.2 (ATP-sensitive potassium channel) or TRPM4 (ATP- and calcium-sensitive non-selective cation channel) (Chen et al., 2003). Interestingly, SUR1-TRPM4 is expressed in injured tissue, but not under physiological conditions (Chen and Simard, 2001; Chen et al., 2003). In human patients with post-traumatic brain contusions, overexpressed SUR1 was found in both astrocytes and microglia, and overexpressed Kir6.2 was observed in astrocytes (Martinez-Valverde et al., 2015; Castro et al., 2019). Additionally, upregulated SUR1, TRPM4, and Kir6.2 were discovered in both TBI patients and a CCI model of TBI (Gerzanich et al., 2019). ATP binding to SUR1-TRPM4 blocks its activity. Thus, ATP depletion leads to channel opening with consequential cation (predominantly Na+) influx, cell depolarization, and cellular swelling (Chen and Simard, 2001). Stokum et al. (2018) demonstrated an assembly of the SUR1-TRPM4-AQP4 complex in a murine model of brain edema using cerebellar cold injury (Stokum et al., 2018). Under physiological conditions, AQP4 creates a complex with other Na+ ion channels, such as TRPV4 (transient receptor potential cation channel subfamily V member 4) (Benfenati et al., 2011) or Na+/K+ ATPase (Illarionova et al., 2010). In the pMCAo (permanent middle cerebral artery occlusion) model of cerebral ischemia, loss of the AQP4-TRPV4 complex in double knockout mice leads to a reduction in the cytotoxic edema/ischemic lesion during the ischemic acute phase (Sucha et al., 2022). Overall, research in AQP4 and its complexes imply, that AQP4 and other channels, such as TRPV4 or TRPM4, may play a crucial role in cellular swelling and the extent of edema development in pathological states associated with AQP4/TRPV4/TRPM4 overexpression (Chmelova et al., 2019; Gerzanich et al., 2019; Sucha et al., 2022).
Following TBI, upregulated SUR1-TRPM4 and AQP4 assemble as a heteromultimeric water/ion channel complex, where SUR1-TRPM4 activity generates an osmotic pressure and causes water influx via AQP4 (Stokum et al., 2018; Figure 4). Interestingly, SUR1-Kir6.2 works contrarily to the SUR1-TRPM4 channel upon ATP depletion; activation of SUR1-Kir6.2 is followed by an outflux of K+ and hyperpolarization of the astrocytic membrane (Simard et al., 2012). To the best of our knowledge, there are no published studies explaining how TBI causes SUR1-Kir6.2 upregulation and which consequences it may cause, but some information is available from ischemia research. In the ischemic pMCAo model, SUR1-Kir6.2 was shown not to participate in cellular swelling while the SUR1-TRPM4 activity contributed to cytotoxic edema development (Woo et al., 2020). Additionally, some articles suggest the contribution of SUR1-Kir6.2 in a better outcome after hypoxia/ischemia (Yamada and Inagaki, 2005; Li et al., 2013).
Furthermore, the microglial immune response to TBI contributes to edema by the induction of AQP4 overexpression. Necrotic neurons from damaged tissue release HMGB1 (high-mobility group box protein 1) which in turn activates microglial TLR4 (Toll-like receptor 4). This event triggers a release of interleukin-6 (IL-6) from microglia, and as a result, it increases astrocytic AQP4 expression (Laird et al., 2014).
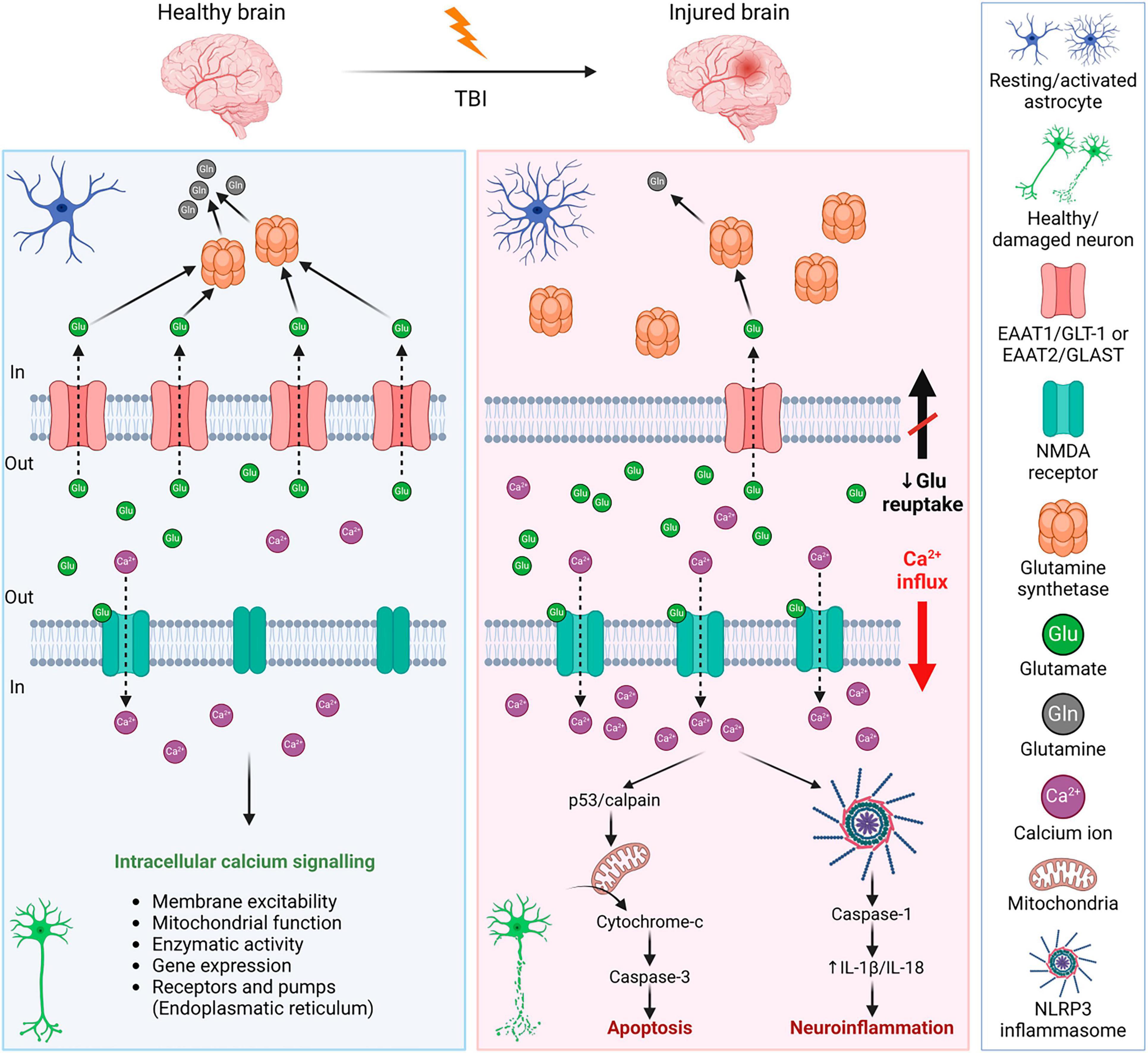
3.2 ExcitotoxicityExcitotoxicity is the process of exaggerated activation of the neuronal amino acid receptors (e.g., NMDA, AMPA, or KA receptors) by their excessive exposure to neurotransmitters, such as glutamate. Consequently, the influx of extracellular Ca2+ into neurons occurs, where Ca2+ may eventually trigger apoptotic signals through calpain mediating p53 induction and following caspase3-dependent neuronal apoptosis (Sedarous et al., 2003; Figure 5). Moreover, the Ca2+ influx induces neuroinflammation via NLRP3 (NLR family pyrin domain containing 3) and caspase-1. NLRP3 inflammasome is an intracellular sensor that detects microbial motifs and endogenous danger signals, providing a fast immune response (Kelley et al., 2019; Johnson et al., 2023). It is a complex composed of three protein subunit types: NLRP3 (a sensor protein), ASC (the adapter protein apoptosis-associated speck-like protein containing a CARD; caspase recruitment domain), and caspase-1. Besides altered calcium signaling, NLRP3 inflammasome is activated by other signals after TBI, including ionic changes (e.g., potassium and chloride efflux), or presence of extracellular ATP and ROS (O’Brien et al., 2020). After activation, NLRP3 inflammasome allows self-cleavage of pro-caspase-1 to active caspase-1, which consequently produces IL-1β or IL-18 (Johnson et al., 2023). The overexpression of IL-1β and IL-18 is followed by neuroinflammation that can lead to neuronal injury, apoptosis, or necrosis (Dong et al., 2009; Abdul-Muneer et al., 2017). Activation of NLRP3 inflammasome can also lead to cell death via pyroptosis (O’Brien et al., 2020; Figure 5).
Figure 5. Mechanisms of glutamate excitotoxicity after traumatic brain injury—a comparison with a healthy brain. Left panel: In a healthy brain, the excess glutamate is cleared by astrocytes, where glutamate is converted into glutamine by glutamate synthetase. It is a strictly balanced environment to prevent neuronal death and neuroinflammation. Right panel: After TBI, the concentration of glutamate in the extracellular space increases due to disrupted BBB, or reduced glutamate reuptake due to the decreased expression of astrocytic glutamate transporters. Increased activation of neuronal NMDA receptors by glutamate binding evokes an influx of Ca2+ into the cells. Excessive Ca2+ triggers apoptotic signals leading to cell death and a release of IL-1β that induces immune response. For further details see text. NMDA receptor, N-methyl-D-aspartate receptor; IL-1β, interleukin 1β; BBB, blood-brain barrier; EAAT1/2, excitatory amino acid transporter 1/2; GLT-1, glutamate transporter 1; GLAST, glutamate aspartate transporter; NLRP3, NLR family pyrin domain containing 3. Created with BioRender.com.
After TBI, levels of extracellular glutamate increased in both patients and experimental models of TBI (Palmer et al., 1993; van Landeghem et al., 2001; Chamoun et al., 2010; Sowers et al., 2021). The excess glutamate contributes to secondary injury and increases the extent of the damage. There are different causes contributing to the elevated extracellular concentration of glutamate, which include increased release (disrupted BBB, dysregulated exocytosis) or decreased reuptake by glutamate transporters. Through glutamate transporters, both astrocytes and microglia participate in glutamate clearance. In the brain, there are several subtypes of glutamate transporters; glutamate transporters EAAT1/GLT-1 and EAAT2/GLAST are predominantly astrocytic, but they are present in microglia as well. Other glutamate transporters (EAAT3-5) are predominantly neuronal. In human patients with TBI, a decreased expression of EAAT1/GLT-1 and EAAT2/GLAST was observed (van Landeghem et al., 2006; Beschorner et al., 2007), which supports the idea of the involvement of glutamate transporters in the tissue damage and final outcome after TBI. Therefore, many studies have focused on glutamate metabolism and glial glutamate transporters.
Lehmann et al. (2009) used cultured cortical astrocytes to study the influence of high glutamate levels on the expression of glutamate transporters and glutamine synthetase (GS), which converts transported glutamate into glutamine. In their study, astrocytic expression of both EAAT1/GLT-1 and EAAT2/GLAST decreased in high glutamate medium (0.5–20 mM) via glutamate receptor-independent mechanism. On the contrary, an increase in GS expression was induced by higher levels of glutamate (≥1 mM) (Lehmann et al., 2009). A significant decrease in the protein expression of glutamate transporters was reported in the ipsilateral cortex during the acute phase following CCI in rats (Rao et al., 1998). In this model, ramified microglia with EAAT2/GLAST expression were present as early as 2 h post-TBI. From 4 up to 72 h after CCI, microglia expressing EAAT1/GLT-1 and EAAT2/GLAST were present in all observed areas, including the cortex, hippocampus, and lateral thalamus (van Landeghem et al., 2001). Yi et al. (2005) studied splice variants GLT-1v and GLT-1α in a rat model of lateral FPI. They observed a significant loss of GLT-1v (c-terminal splice variant) in the cerebral cortex (6–24 h post-TBI) and a transient decrease in the hippocampus and thalamus (6 h post-TBI). Interestingly, there was no significant change in GLT-1α in the cortex, but an increase was observed in the hippocampus (6–24 h post-TBI) (Yi et al., 2005). Additionally, it was found that GFAP (which is overexpressed after TBI) may modulate astrocytic glutamate transport in a region-dependent manner and be involved in EAAT2/GLAST trafficking (Hughes et al., 2004).
3.3 NeuroinflammationFollowing TBI, the immune response is a natural process, which occurs within minutes post-injury. Although neuroinflammation is a necessary reaction to injury, its persistence causes long-term complications in TBI patients. Chronic neuroinflammation not only contributes to tissue damage but there is also growing evidence showing it as a main feature of other brain pathologies, such as dementia or Alzheimer’s disease (AD) (Eikelenboom et al., 2010; Perry et al., 2010). TBI itself is therefore considered a risk factor for AD and dementia development later in life (Plassman et al., 2000; Fleminger et al., 2003; Li et al., 2017), as persistent microgliosis is a common factor of these conditions.
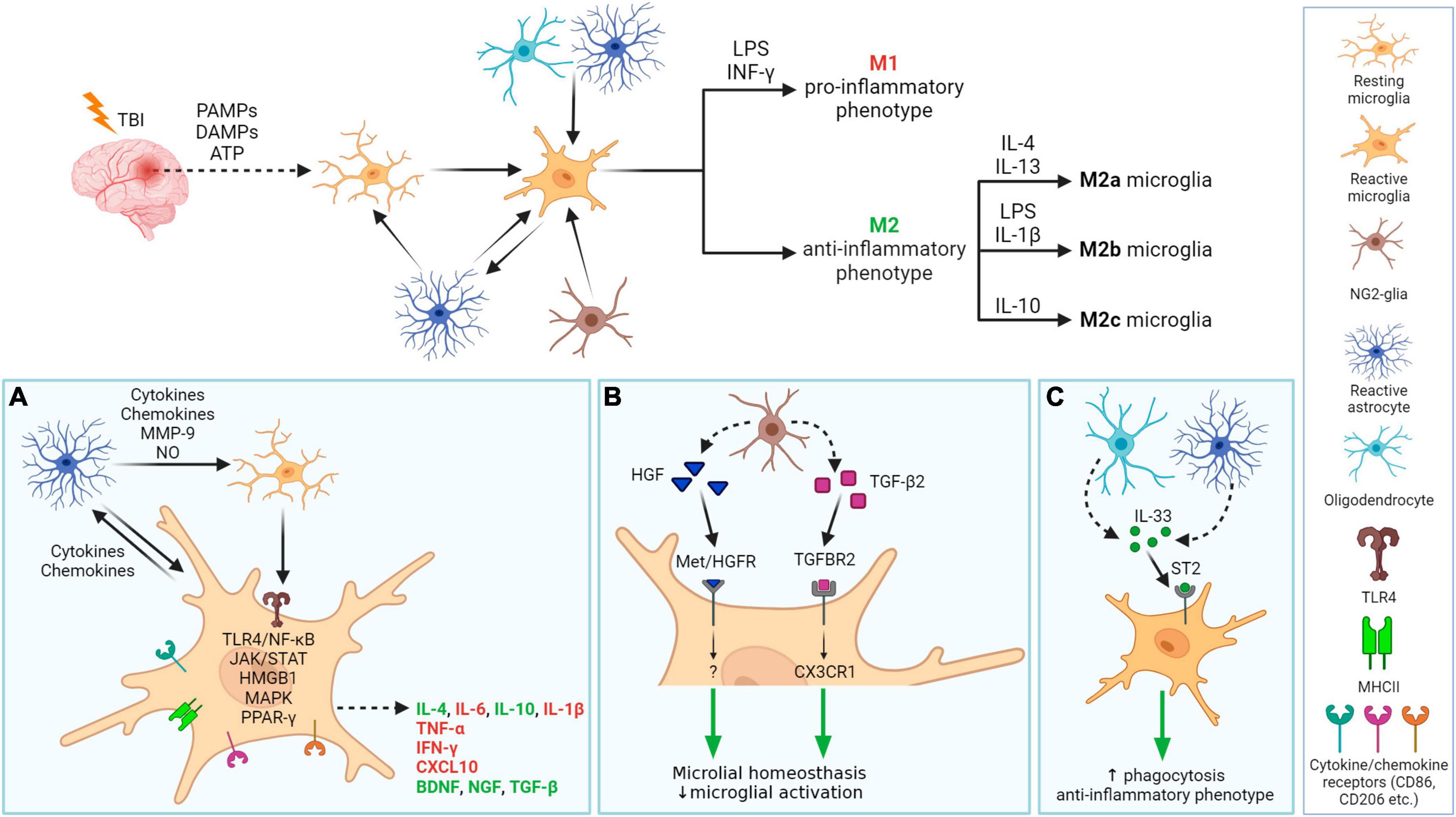
Both microglia and astrocytes are involved in neuroinflammation, and their interplay is essential (Figure 6). Following TBI, microglia are activated by PAMPs (pathogen-associated molecular patterns) or DAMPs (damage-associated molecular patterns) released from damaged tissue (Jassam et al., 2017; Johnson et al., 2023). The process of microglial activation is complex and heterogeneous, depending on the severity, type of injury, and the extent of the damaged area and structure of the brain (Shields et al., 2020). In general, microglia express receptors recognizing cytokines, chemokines (e.g., CD86, CD206), and after activation, also major histocompatibility complex class II (MHCII). Microglia are activated by pro-inflammatory cytokines such as interferons (INF-γ), interleukins (e.g., IL-6), or tumor necrosis factor (TNF-α). Additionally, a fast microglial response is supported by ATP, released from damaged tissue, and by activated astrocytes (Davalos et al., 2005). Reactive astrocytes also contribute to microglial activation by producing cytokines, chemokines, nitric oxide (NO), or matrix metalloproteinase 9 (MMP-9). Furthermore, astrocytes, together with oligodendrocytes, secrete IL-33, which promotes the recruitment of microglia and macrophages post-TBI (Wicher et al., 2017). The production of inflammatory cytokines is mediated by TLR4 via TLR4/NF-κB pathway. During TBI, upregulation of TLR4 can be observed in hippocampal astrocytes and neurons and its depletion suppresses the production of pro-inflammatory cytokines IL-6, IL-1β, and TNF-α. Other pathways involved in signaling during neuroinflammation include JAK/STAT (Janus kinase/Signal transducers and activators of transcription), HMGB1, MAPK (Mitogen-activated protein kinase), or PPAR-γ (peroxisome proliferation-activated receptor γ). They are discussed in more details below.
Figure 6. Neuroinflammation processes in damaged tissue after traumatic brain injury with a focus on glial cells. After TBI, PAMPs, DAMPs, and ATP are released from damaged tissue, which together with molecules (cytokines, chemokines) released by astrocytes, activate microglia. Activated microglia migrate into the damaged area and release pro- or anti-inflammatory factors. Microglial phenotype M1 and M2 (M2a-c resp.) phenotypes can be specifically activated. Other glial cells also have a role in neuroinflammation (A–C). (A) In damaged tissue, cellular communication between activated astrocytes and resting or activated microglia occur. Activated microglia receive signals via their receptors that trigger one of the cascades, e.g., ligand binding to TLR4 induces NF-κB dependent pathway that evokes the release of IL-6, IL-1β, or TNF-α.. Released molecules can have pro-inflammatory (red) or anti-inflammatory properties (green). (B): Microglial neuroinflammation activity can be regulated by NG2-glia, by releasing HGF of TGF-β2. (C) Both astrocyte and oligodendrocytes release IL-33, which promotes microglial anti-inflammatory phenotype and phagocytic ability. PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; HGF, hepatocyte grow factor; TGF-β2, transforming grow factor β2; INF-γ, interferon γ; LPS, lipopolysaccharide; IL, Interleukin, e.g., IL-4; TLR4, toll-like receptor 4; MHCII, major histocompatibility complex class II; CD, cluster of differentiation, e.g., CD86; ST2, receptor suppression of tumorigenicity 2, also known as IL1RL1 or IL33R; interleukin-1 receptor-like 1, or interleukin-33 receptor; Met/HGFR, hepatocyte growth factor receptor encoded by the MET gene; TGFBR2, transforming growth factor beta receptor 2; CX3CR1, C-X3-C motif chemokine receptor 1; MMP-9, matrix metalloproteinase 9; NO, nitric oxide; TNF-α, tumor necrosis factor α; CXCL10, C-X-C motif chemokine ligand 10; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor; NF-κB, nuclear factor κB; JAK/STAT, janus kinase/signal transducers and activators of transcription; HMGB1, high-mobility group box 1; MAPK, mitogen-activated protein kinase; PPAR-γ, peroxisome proliferator-activated receptor γ. Created with BioRender.com.
Galectins act as master regulators in the inflammatory response associated with neurodegenerative disease development (Soares et al., 2021). Yip et al. (2017) found that activated microglia release galectin-3 as a TLR4 ligand, crucial for the full microglial response to proinflammatory stimuli, like lipopolysaccharide (LPS) (Yip et al., 2017). Furthermore, galectin-3 influences the inflammatory response to α-synuclein (α-syn) in microglial cells and act as a natural ligand of TREM2 (Triggering receptor expressed on myeloid cells 2) receptor involved in AD pathogenesis (Boza-Serrano et al., 2014; Qin et al., 2021). The role of galectin-3 has been intensively studied in ischemia and tumorogenesis, where it was shown to activate or modulate other signaling molecules and regulators of cellular processes such as interleukin kinase or tyrosine kinases (Wesley et al., 2013; Porebska et al., 2021). However, several studies indicate a significant early increase in galectin-3 expression also in various trauma models, including experimental TBI (Natale et al., 2003; Venkatesan et al., 2010). Yip et al. (2017) discovered significant galectin-3 expression in mouse cerebrospinal fluid (CSF) 24 h post-injury, likely stemming from a disrupted BBB (Yip et al., 2017). Galectin-3-dependent-TLR4 activation may contribute to prolonged microglia activation and sustained brain inflammation (Burguillos et al., 2015). However, another study revealed a positive correlation between human plasma galectin-3 levels and higher GCS scores (Shen et al., 2016). These findings suggest the potential role of galectin-3 as a biomarker in TBI, indicating a need for further exploration for clinical applications. In contrast, the other member of galectin family, galectin-1, suppresses activation markers, proinflammatory cytokines, and inducible nitric oxide synthase (iNOS) expression in microglia in inflamed CNS tissues, while its absence prompts classical microglial activation (Aalinkeel and Mahajan, 2016).
Following activation, microglia undergo polarization—they change their morphology into larger rounded ameboid cells, proliferate and migrate into the damaged region (Caplan et al., 2020). Activated microglia also start to release factors with pro-inflammatory/anti-inflammatory functions. The basic microglial phenotypes after activation can be distinguished into two types–the M1 type and the M2 type. M1 type is considered a pro-inflammatory phenotype, preferably producing pro-inflammatory compounds such as cytokines TNF-α, INF-γ, IL-1β, chemokines (e.g., C-X-C motif chemokine ligand 10, CXCL10), or ROS. Alternatively, the neuroprotective M2 phenotype produces anti-inflammatory molecules, such as interleukin IL-10 (which can also be expressed by the M1 phenotype, but to a lower extent) (Mills et al., 2000; Ziebell and Morganti-Kossmann, 2010). The M2 phenotype has 3 subtypes: M2a, M2b, and M2c, which differ in function, activation factor, and expression of specific markers (Zhang et al., 2022a). Microglial phenotype can be specifically activated: IFN-γ and LPS stimulate M1 phenotype; IL-4 and IL-13 trigger M2a phenotype; M2b phenotype is the result of LPS or IL-1β activation and IL-10 triggers M2c phenotype (Figure 6). Each of these phenotypes has a specific proteomic profile (Xu et al., 2017; Vergara et al., 2019). Notably, transcriptomic analysis of activated microglia revealed the limitations of these classifications—activated microglia rarely fit the classification perfectly; usually a mixed microglial population is observed. Furthermore, microglial gene expression is dynamic and time-dependent. Izzy et al. (2019) demonstrated the reduced ability of microglia to sense tissue damage in the early stage of TBI, which in a later stage (14 days post-injury, dpi) began to transfer into a specialized inflammatory state, with changes of IL-4, IL-10, and IFN-γ gene expression within 14–60 dpi (Izzy et al., 2019). Due to this diversity, Paolicelli et al. (2022) oppose dual nomenclatures, such as M1/M2 or pro- vs. anti-inflammatory microglia. Instead of restricting to these types, the authors advise using descriptions of phenotypes and transcriptome (Paolicelli et al., 2022).
Until recently, it was thought that only astrocytes and microglia contribute to neuroinflammation. However, recent studies have revealed the important role of NG2-glia in the brain immune system as well as its involvement in neuroinflammation (Figure 6). Nakano et al. (2017) reported that the elimination of NG2-glia activates the IL-1β pro-inflammatory pathway resulting in defects of hippocampal neurons due to neuroinflammation. Furthermore, NG2-glia-expressed hepatocyte growth factor (HGF) acts as a regulator of the neuroinflammation level (Nakano et al., 2017). According to Zhang et al. (2019), transforming growth factor beta receptor 2 (TGF-β2) expressed by NG2-glia regulates the microglial chemokine receptor CX3CR1 (C-X3-C motif chemokine receptor 1) via increased phosphorylation of the transcriptional modulator mothers against decapentaplegic homolog 2 (SMAD2) and therefore controls microglial homeostasis. In any case, the deficiency of NG2-glia in the Parkinson’s disease (PD) mouse model contributed to neuroinflammation (Zhang et al., 2019). These studies suggest that NG2-glia may have a role in neuroinflammation regulation in both physiological and pathological states.
3.4 Cell death, demyelination, and white matter degradationCellular death typically accompanies TBI and was observed in both TBI patients and experimental models. Cellular death was documented in several brain regions, including the cortex, hippocampus, and thalamus (Clark et al., 1997; Conti et al., 1998; Fox et al., 1998). Apoptosis following TBI is triggered by a caspase-dependent pathway (Yakovlev et al., 1997; Clark et al., 2000) or a caspase-independent pathway, where apoptosis is induced by several mitochondrial proteins, e.g., mitochondrial apoptosis-inducing factor AIF (Zhang et al., 2002). Apoptosis is region dependent, with an early response in white matter (12 h post-TBI) and a delayed apoptosis peak in the thalamus (2 weeks post-TBI) (Conti et al., 1998). Neurons and oligodendrocytes in white matter are more vulnerable to apoptosis following TBI than the rest of the cells in the brain. Loss of oligodendrocytes is supplied by NG2-glia proliferation, which can be observed even months after injury (Dent et al., 2015), and the differentiation into oligodendrocytes can be driven by myelin damage (Hill et al., 2014). Johnson et al. (2013a) found ongoing white matter degradation in the corpus callosum, and persistent neuroinflammation in patients 3 months post-injury (Johnson et al., 2013a). Flygt et al. (2016) observed oligodendrocyte cell death and an increase in the number of oligodendrocyte progenitor cells/NG2-glia in patients with moderate to severe TBI (Flygt et al., 2016). Oligodendrocyte cellular death is caused by several factors—the microglial release of inflammatory cytokines, such as TNF-α or IFN-γ, higher oxidative stress, increased levels of extracellular ATP and/or excess of glutamate, which commonly occurs in TBI tissue. Interestingly, Lotocki et al. (2011) demonstrated a positive effect of hypothermia on TBI-induced oligodendrocyte loss—a decrease in temperature after injury for 4 h led to suppression of caspase-3 activity, which consequently enhanced oligodendrocyte survival (Lotocki et al., 2011).
Since oligodendrocytes produce myelin, a decrease in the amount of myelin after TBI could be the result of oligodendrocyte apoptosis. Both white matter loss and demyelination can be detected in various brain regions. Damaged areas with atrophy include the corpus callosum (Johnson et al., 2013a; Green et al., 2014; Briggs et al., 2016), hippocampus (Green et al., 2014) pons or cerebellum (Spanos et al., 2007). TBI activates IKK/NF-κB (IKK, inhibitory-κB kinase activates NF-κB by phosphorylating IκBα, the inhibitory subunit of NF-κB) signaling in oligodendrocytes, which induces their senescence and is followed by disturbed myelination (Schlett et al., 2023). Mahoney et al. (2022) used magnetic resonance imaging to demonstrate that activated intracortical demyelination has a distinct spatial profile in comparison to the acute phase. Additionally, the impact of TBI-related demyelination manifests worse outcomes than age-related demyelination, where temporal, cingulated, and insular regions showed a higher degree of myelin loss (Mahoney et al., 2022). Demyelination is spontaneously followed by remyelination; this process has a protective effect ensuring axonal survival (Irvine and Blakemore, 2008). However, the remyelination process is limited by inflammation on the site of the demyelinated lesions and inadequate precursor differentiation.
Microglia, together with macrophages, remove dying cells, but the alleviation of inflammatory responses may also enhance apoptotic processes. According to Wang et al. (2020), microglial depletion promoted neurite growth and reduced the total neural apoptosis after the FPI model of injury (Wang et al., 2020). The microglial pro-inflammatory response can directly trigger apoptosis of oligodendrocytes; conversely the anti-inflammatory M2 phenotype of microglia support remyelination (Miron et al., 2013), therefore a balance between M1 and M2 phenotype seems to be essential for the remyelination process. Blocking of microglial NHE1 (Sodium-hydrogen antiporter 1), the protein responsible for NADPH oxidase and cytokine secretion in pro-inflammatory microglia, resulted in a reduced inflammatory response, and consequently promoted oligodendrogenesis and supported remyelination in TBI tissue (Song et al., 2022).
3.5 Reactive gliosis and scar formationThe formation of a scar following TBI is considered a defense mechanism, preventing the propagation of the tissue damage and the spread of toxic metabolites; however, it also prevents axonal growth leading to impaired neural function recovery (Silver and Miller, 2004; Burda and Sofroniew, 2014; Karve et al., 2016).
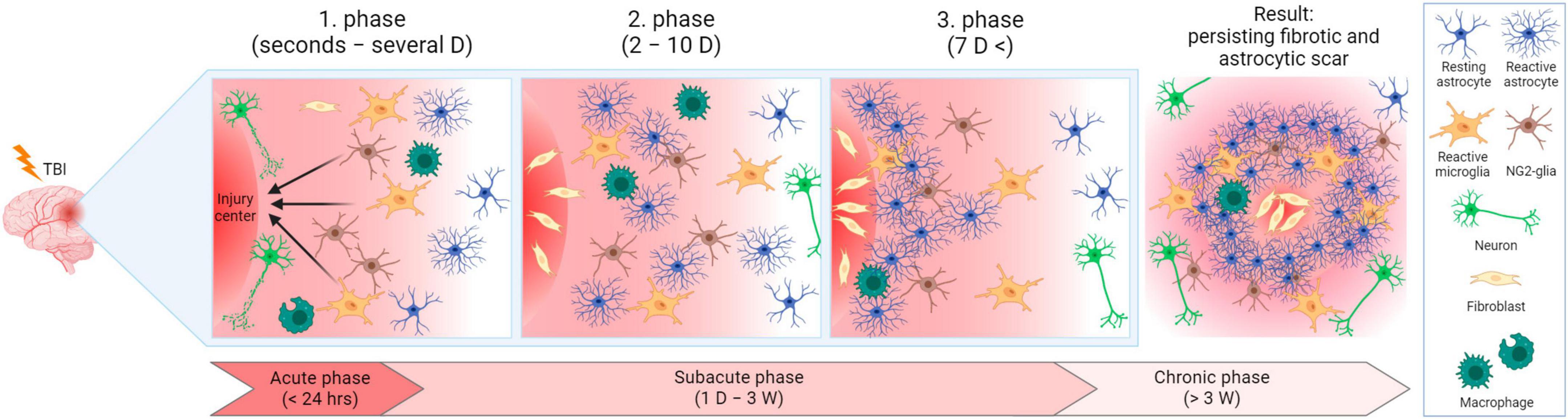
After TBI, it is plausible to divide the ensuing events into three phases, while the processes of these phases can overlap (Burda and Sofroniew, 2014; Figure 7). In the first phase (seconds to hours after injury, prolonged up to several days), trauma evokes acute cell death in the injury center. Immune cells are activated and recruited, including microglia. Microglia and NG2-glia migrate toward the damaged area, astrocytes do not migrate, but swell and can eventually become reactive. During the second phase (2–10 days after injury), cells, including scar-forming astrocytes, proliferate. During this phase, a lesion core containing fibroblasts, pericytes, or inflammatory cells, starts to form. The core is surrounded by a layer of reactive astrocytes separating the core and the peri-lesion perimeter, which contains reactive glial cells, and gradually transitions to healthy tissue. Tissue remodeling appears in the third phase (which can occur 7 dpi) and the lesion matures. The result is a persisting fibrotic and astrocytic scar. Tissue remodeling is also present in the peri-lesion perimeter and can persist for months post-TBI (Burda and Sofroniew, 2014).
Figure 7. Lesion development after traumatic brain injury in time. In the acute phase, cells are damaged in the injury center, and immune cells are activated and recruited. Microglia and NG2-glia migrate toward the injured area and astrocytes become reactive. In the subacute phase, cells (including scar-forming astrocytes) proliferate. A layer of reactive astrocytes surrounds a forming lesion core. During the transition between the subacute and chronic phases, tissue remodeling and lesion maturation occur, resulting in permanent fibrotic and astrocytic scars. D, day/days; W, week/weeks. Created with BioRender.com.
However, a mild diffused injury may result in gliosis without scar core formation (Shandra et al., 2019). In various articles, the term “glial scar” or “astroglial scar” is used for the whole lesion. Therefore, it was suggested not to use these terms because astrocytes are not mesenchymal or stromal cells, which form scars in other damaged tissues (e.g., skin or heart). Hence, “scar formation” occurring in the brain after TBI does not correspond to the widely used terminology from other research areas (Sofroniew, 2020). In contrast, other studies use the term “glial scar” only to describe the glial border of the scar since glial cells are not the main compounds of the lesion (Adams and Gallo, 2018).
NG2-glia together with microglia are the first glial cells to react to the injury, though microglia extend their processes faster than NG2-glia (Hughes et al., 2013). During injury, NG2-glia re-enter the cell cycle, which in combination with the shortening of the G1 phase, results in their proliferation and an increase of the NG2-glia population (Simon et al., 2011). OPCs, which belong to the NG2-glia population, are recruited via TLR-2/CXCR3 pathway signaling to the lesion site, where they differentiate into oligodendrocytes (Sanchez-Gonzalez et al., 2022). However, part of the NG2-glia form the glial border together with astrocytes and microglia, without any differentiation (Levine, 1994). Since the reduction of the NG2-glia population delays wound closure, it was suggested that accumulation of NG2-glia on the lesion site also has a positive effect on tissue regeneration (von Streitberg et al., 2021).
By releasing signaling molecules, including cytokines (TNF-α, IL-1β, IL-6) or IGF-1 (Insulin-like growth factor-1, in spinal cord injury), microglia stimulate the transformation of astrocytes into reactive astrocytes, which are a key element of th
留言 (0)