
記住我



Ischemic stroke has been studied extensively due to its pervasive nature on patients’ quality of life and economic burden on the public health care system, nevertheless, the current available treatment provides less than satisfactory results (French et al., 2016). In clinical medicine as well as in the experimental research, close attention is usually given to the region with the greatest destruction, the ischemic core, as well as to the tissue with recovery potential, the penumbra (Saver, 2017). This focus on the parts that are affected the most seriously is understandable since those areas undergo the most noticeable alterations. However, the understanding of ischemic mechanisms would not be complete if we only considered the most severely damaged regions, since the seemingly undamaged brain regions may also fall victim to the secondary injury. This process was assigned as exo-focal neuronal death (Zhao et al., 2002) (although neurons are certainly not the only ones affected, see below) or more generally diaschisis, describing the phenomenon where transient malfunction of a distant area occurs as a result of local brain injury (Yang et al., 2013). Ischemic stroke is not the only condition linked to diaschisis – it has been described in cases of epilepsy, migraine, encephalitis, brain tumors and traumatic brain injuries (Paradowski and Pawlik, 2005; Poretti and Boltshauser, 2012), i.e., generally with pathologies associated with inflammation, glia activation, spreading depolarization, apoptosis, oxidative stress and ionic imbalance in the intracellular or extracellular space (ECS).
Even though the site of injury is demarcated by a barrier comprised of cells and fibrous material (Wang et al., 2018) partially preventing the damage from spreading, ischemia also poses a tremendous burden on the healthy surrounding tissue, since not all defensive mechanisms can be engaged at once (Shi et al., 2019). For instance, depolarization elicited in the penumbra do not dissipate at the border with healthy tissue but propagate further (Andrew et al., 2022). The effect of ischemia can then be manifested in the remote areas in the similar way as in the core only in a less profound intensity or reversed manner (Arvidsson et al., 2001; Li et al., 2020). Nevertheless, the changes in remote areas have one thing in common – they appear with a delay after the initial reaction in the core (Zhang et al., 2012; Li et al., 2020). An assessment of whether the damage in remote areas may participate in the final behavioral or cognitive deficit after a stroke attack may be rather difficult for several reasons. First, the association of an injury to a specific brain region with a neurological deficit is complex, and the regional damage may not correspond with the typical behavioral and/or cognitive outcome (Zhou et al., 2013). Second, the disturbance of mental functions can be orchestrated by different parts of the brain, just as normal body functions are not controlled solely by one brain structure (Poldrack, 2010). Moreover, new areas can adopt the role of the damaged region after stroke (Gerloff et al., 2006). Thus, it is reasonable to assume that the state of remote regions matters; certainly the evidence supports this presumption. Several studies have reported a significant correlation between a decrease in regional blood flow and metabolism in the remote cerebellum with clinical stroke scales (Liu et al., 2007; Szilagyi et al., 2012; Shinohara et al., 2017; Wang et al., 2020; Chen et al., 2022). Interestingly, Takasawa M. et al. (2002) were able to obtain such results in the subacute post-ischemic stage, when the changes in remote areas start to manifest, but not in the acute phase (Takasawa M. et al., 2002). Alternatively, we can look at the problem from another point of view: the hippocampus is usually spared from ischemic insult, as the blood supply is not provided by the middle cerebral artery (MCA) (Rusinek et al., 2011), which is a blood vessel frequently occluded during a stroke (Rovito et al., 2021). Therefore, the hippocampus is often considered a region remote from the ischemic core (Gulyaeva et al., 2021). However, depression and dementia are common post-stroke complications that are associated with impaired function of the hippocampus, which could be regarded as intact at first sight (Onufriev et al., 2021). The distant areas may thus play an important role in the clinical outcome. Moreover, in contrast to the rapidly damaged core, we can take advantage of the delayed exo-focal reaction and preserve the still intact remote tissue by using a suitable intervention (Kidani et al., 2020).
In addition to neurons, glial cells are also affected and responsive to ischemic injury (Mihailova et al., 2023). Due to their multiple functions and the large number of released cytokines/chemokines, glial cells often have a “dual face” as they can play an irreplaceable role in mitigating/preventing tissue damage, but they also exacerbate inflammation and excitotoxicity (Pekny et al., 2014; Quincozes-Santos et al., 2021). Therefore, glia-oriented therapies aimed at the detrimental functions of glia, that can intensify or propagate ischemic injury, progressively prevail over inefficient neuron-centered approaches in the preclinical phase [for review see (Hernandez et al., 2021)]. The glia-related mechanisms, such as neuroinflammation, reversed glutamate uptake, extracellular matrix (ECM) remodeling, disruption of myelin sheets and calcium waves are already generally accepted as contributors of ischemic injury, spreading from the core to the penumbra. However, their involvement in the propagation of the ischemic insult into the remote areas remains hypothetical and has yet to be fully elucidated. In this review, using the research data published in the last 20 years, we present comprehensive information regarding the impact of focal ischemia on brain tissue outside of the penumbra. We discuss in particular the post-ischemic reaction of neurons, glial cells and ECM in various brain regions and the possible therapeutic approaches. Supplementary Table 2 provides a concise summary.
2 Glial cells and their role in ischemic injuryAlthough glial cells were originally described by Virchow’s and other early studies as purely supportive elements in the brain (Chvatal and Verkhratsky, 2018), years of intensive research revealed their multifaceted function in development and physiological conditions, as well as in various central nervous system (CNS) injuries and diseases, including ischemia (Verkhratsky, 2007; Allen and Lyons, 2018; Mihailova et al., 2023). Here, we briefly introduce the most well known and most studied glial cell types in ischemia - astrocytes, microglia, oligodendrocytes and NG2-glia. The following chapters will discuss more detailed information, focusing on the structural/functional alterations and localization of individual glial cell types in post-ischemic tissue and the distinct remote areas, and their participation in ischemic damage and propagation.
Astrocytes, the most prevalent type of glial cells, as an important component of the blood-brain barrier (BBB), can improve nutritional support for neurons by regulating the capillary blood flow and releasing ketone bodies that supply energy. In addition, the astrocytic uptake of excitotoxic substances, mainly glutamate, by transporting proteins present in their plasma membrane, helps to restrict the spread of damage to the surrounding environment (Hernandez et al., 2021). Furthermore, astrocytes also help maintain a stable pH and ionic homeostasis by removing the protons and potassium released in the ECS during neural activity (Gradisnik and Velnar, 2023). Ischemia-induced injury triggers astrocyte activation, including changes in protein expression (e.g., upregulation of glial fibrillary acidic protein, GFAP), and morphological changes, such as branching levels and length of processes (Pekny and Nilsson, 2005). These morphological changes are dependent on the distance from the infarction area (Li et al., 2022). Based on their gene expression and consequent role, activated astrocytes can be divided into 2 main types: A1 (inflammation-induced, pro-inflammatory) and A2 astrocytes (ischemia-induced, anti-inflammatory) (Zamanian et al., 2012; Liddelow et al., 2017). The A1 type is considered harmful, releasing pro-inflammatory mediators (e.g., interleukin IL-6, or tumor necrosis factor α, TNF-α) and acting detrimental to synapses. The A2 type release anti-inflammatory compounds and growth factors, such as brain-derived neurotrophic factor (BDNF) and promote the survival and growth of neurons (Liddelow and Barres, 2017; Xie and Liu, 2023). However, the development of advanced transcriptomic analyses, including single-cell RNA sequencing, has led to an accumulation of evidence that there are multiple types of reactive astrocytes. For example, in post-ischemic penumbra, 7 subgroups of astrocytes were identified (Guo et al., 2021).
Astrocytic glutamate uptake in ischemia can be reversed and thus enhances excitotoxic damage (Gouix et al., 2009). Moreover, ischemia-evoked astrocytic swelling is accompanied by compensatory ECS shrinkage that further increases the concentration of potentially toxic agents (Sykova, 2004; Lafrenaye and Simard, 2019). In addition, astrocytes contribute to the BBB breakdown by detaching their endfeet from capillaries and producing substances that promote blood vessel permeability [e.g., vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs), nitric oxide (NO), and endothelin-1] (Zhang et al., 2020). Structural alterations in reactive astrocytes and overproduction of ECM hinder the flow of growth hormones and neuroactive substances across the ECS, impairing their potential regeneration as well as extrasynaptic intracellular communication (Vargova and Sykova, 2014; Wang et al., 2018). Moreover, astrocytes coupled by gap-junctions create a syncytium, which, by the spread of calcium waves and ATP release, enables the so-called gliotransmission and distant neuro-glia communication. Under ischemic conditions, these mechanisms allow the propagation of the injury into the neighboring tissue and may thus affect the cells in remote areas (Verkhratsky, 2007).
Microglia are mainly “cleaning agents”, that eliminate potentially harmful substances as well as dysfunctional synapses and thus mediate tissue remodeling. In addition, in later periods of post-ischemic regeneration, they release a variety of neuroprotective factors (Xu S. et al., 2020). Both microglia and astrocytes produce pro-inflammatory cytokines when activated by ischemia, and the dysregulated inflammatory response can worsen the functional and tissue damage in the ischemic brain (Xu S. et al., 2020). Activation of microglia includes a change in gene expression [e.g., upregulation of ionized calcium-binding adapter molecule 1 (Iba-1), and a cluster of differentiation 68 (CD68)] as well as microglial polarization (Ito et al., 2001; Perego et al., 2011). Similarly to astrocytes, the phenotype of activated microglia can be either the pro-inflammatory M1 type or the anti-inflammatory M2 type. The M1 phenotype produces inflammatory cytokines and chemokines, such as TNF-α, IL-1β, or interferon-γ (IFN-γ), and promotes neuronal death. The M2 acts as a beneficiary phenotype, releasing anti-inflammatory compounds (IL-10, IL-4, or transforming growth factor TGF-β) and neurotrophic factors (Lee et al., 2014). In the early stages of ischemia, M2 predominates over the M1 type. Later, the microglial phenotype is gradually shifted toward the M1 type in peri-infarct regions (Hu et al., 2012). However, more recent studies using gene expression profiling showed that the division of microglia into only 2 types is not sufficient. Using single-cell RNA-sequencing, Guo et al. (2021) found 14 microglial subgroups, which showed significant variability in expression profiles and uneven distribution between the ischemic middle cerebral artery occlusion (MCAO) and the control group (Guo et al., 2021).
Oligodendrocyte progenitor cells, also known as polydendrocytes or NG2-glia, exhibit a high proliferative and differentiation ability, primarily in myelinating oligodendrocytes under physiological conditions (Zhu et al., 2008; Dimou and Gallo, 2015). Under ischemic conditions, the number of NG2-glia was significantly decreased in the infarct core but significantly elevated in the penumbra (Tanaka et al., 2001). NG2-glia contribute to glial scar formation and wound closure, regulate neuroinflammation and are endowed with a high proliferative ability (Valny et al., 2017). Interestingly, under ischemic conditions, NG2-glia differentiate rather into reactive astrocytes than into oligodendrocytes, as evidenced by the immunohistochemical and electrophysiological properties of glia cells in ischemia (Honsa et al., 2016).
Oligodendrocytes are myelinating cells that sustain and insulate axonal myelin sheaths. Ischemia, accompanied by oxidative stress or excitotoxicity has a detrimental effect on oligodendrocytes, leading to their apoptosis and demyelination, which can have a significant impact on the neurological functions and final outcome of ischemia (Dewar et al., 2003).
Tissue response to ischemic injury comprises several complex mechanisms where glial cells can play pro-active or suppressive roles. These mechanisms include neuroinflammation (Shen et al., 2023), edema (Gu et al., 2022), oxidative stress (Radak et al., 2014), excitotoxicity (Kirdajova and Anderova, 2020), and glial scar formation (Silver and Miller, 2004; Kawano et al., 2012; Manrique-Castano and ElAli, 2021). The vast majority of these processes are considered detrimental, aggravating and expanding the tissue damage. However, the formation of a glial scar stands out among these reactions to ischemic/traumatic insult, as it assumes a dual role in the injured CNS. In the early stages of glial scar formation, reactive astrocytes release BDNF and suppress inflammation, protecting nerve cells from further damage (Rolls et al., 2009). In addition, scar formation also creates a barrier by depositing fibrotic molecules at the injury site, thus impeding neurotoxic substances, peripheral leukocytes and inflammatory signals to enter healthy tissue (Manrique-Castano and ElAli, 2021). Altogether, these actions safeguard neural tissue from propagating traumatic or pathological insults. While contributing to certain tissue protection, glial scar formation has negative consequences as well. They appear especially in later post-ischemic stages and include hindering the reconstruction of the BBB or preventing the promotion of axonal growth. Moreover, due to the release of pro-inflammatory cytokines, glial scar contributes to persistent widespread inflammation, promoting tissue degeneration (Kawano et al., 2012; Zhang et al., 2020; Manrique-Castano and ElAli, 2021). Interestingly, Zbesko et al. (2018) suggested that glial scar does not completely isolate the damaged area from healthy tissue but is partially permeable to toxic compounds contained in extracellular fluid released from the “area of liquefactive necrosis” (Zbesko et al., 2018).
3 Definition of the terms ischemic core, penumbra, and remote areasWithin the first several hours following the ischemic insult, three affected areas can be recognized: (1) the ischemic core; (2) the penumbra; (3) the remote areas.
The core may be defined as a region with a decrease of regional blood flow below 35 % in the grey matter and below 25 % in the white matter (WM) in human brain (Rodriguez-Vazquez et al., 2022). Hartings et al. (2017) claim the core can be best described as an area of persistent depolarization with the regional blood flow failing to reach 5 – 10 ml/100 g/min (Hartings et al., 2017).
The penumbra was initially characterized in a monkey model of stroke as tissue undergoing progressive damage surrounding a uniform central core destined for infarction (Astrup et al., 1977, 1981; Symon, 1980). The “tissue at risk” concept of the penumbra was based on intensive research in experimental animal models of stroke (Ebinger et al., 2009). The development of imaging techniques such as positron emission tomography (PET), computing tomography (CT) and magnetic resonance imaging (MRI) allowed visualization of the penumbra and helped to verify this concept also in humans, with varying core and penumbra definitions according to the used technique (Ermine et al., 2021). The penumbra is also deemed a salvageable area if prompt reperfusion is achieved (Witte et al., 2000). Interestingly, based on numerous studies involving animal models and stroke patients, del Zoppo et al. (2011) have suggested that both the infarct core and the ischemic penumbra exhibit heterogeneity in the early minutes and hours after ischemia. Their model consists of “mini-cores” surrounded by multiple “mini-penumbras”; without intervention, these “mini-penumbras” will be consumed by expanding “mini-cores”, and consequently encompassing larger region of injury (Jones et al., 1981; Tagaya et al., 2001; del Zoppo et al., 2011). In the non-reperfused penumbra, the apoptosis of still viable cells is induced, which leads to the spread of the ischemic core over a period of several hours (Liu et al., 2010; Genova, 2011; Saver, 2017). The crucial roles in the shift from reversible to irreversible tissue damage play spreading depression-like depolarization and excitotoxicity (Hartings et al., 2017; Sueiras et al., 2021; Andrew et al., 2022). This is one of the reasons why some researchers consider the precise division of ischemia-affected parts outdated (van Putten et al., 2021). However, several clinical studies confirmed that rescuing the penumbra in patients with a convenient core/penumbra imaging profile can considerably extend the therapeutical time window for thrombolytic therapy (Ermine et al., 2021).
The remote areas have been defined as regions undergoing tissue transformation, without cellular death (Karetko-Sysa et al., 2011), however, this is not always the case (Uchida et al., 2010; Park et al., 2011). According to some studies, the core is the only area of the affected regions where structural changes occur, whereas the remote structures undergo solely functional alterations (Poretti and Boltshauser, 2012; Yang et al., 2013). Nevertheless, there is a body of research that shows the opposite can be true; readers can find various examples of post-ischemic alterations in cell morphology, and a number of synapses or protein accumulations in this article.
The controversies between the studies arise mostly from the vague and subjective spatial/temporal definition of remote areas. Certainly, to indicate precisely the spatial characteristics of remote areas is almost impossible since it depends on the location, severity and mainly the duration of the vessel occlusion. However, there are some conditions that should be fulfilled in the “proper” remote areas. First of all, the vessels supplying the ischemic and remote areas differ, hence the remote areas are not directly affected by the initial deprivation of the blood flow but may undergo secondary changes or damage due to various processes triggered by the ischemic event (Karetko-Sysa et al., 2011). Second, the remote areas are usually located in a different brain structure than the ischemic core; in the case of the cortex, it should be a different lobe or even contralateral hemisphere (Bonilha et al., 2014). Third, there should exist functional connections between the core and the remote area. The temporal definition is slightly easier as the first changes in cell structure and functions in the core or penumbra can occur within minutes after ischemic onset (cytotoxic swelling), rapidly develop within hours (cell death) and the first week (glia activation and proliferation, ECM alterations, development of glial scar) and stabilize several months after ischemia (permanent glial scar formation or recovery in the case of reperfused penumbra). In contrast, the first changes in the remote areas can be detected within days or even months after stroke. Moreover, these alterations are mostly functional while structural changes in the remote areas are subtler than in the core or penumbra or they may be none.
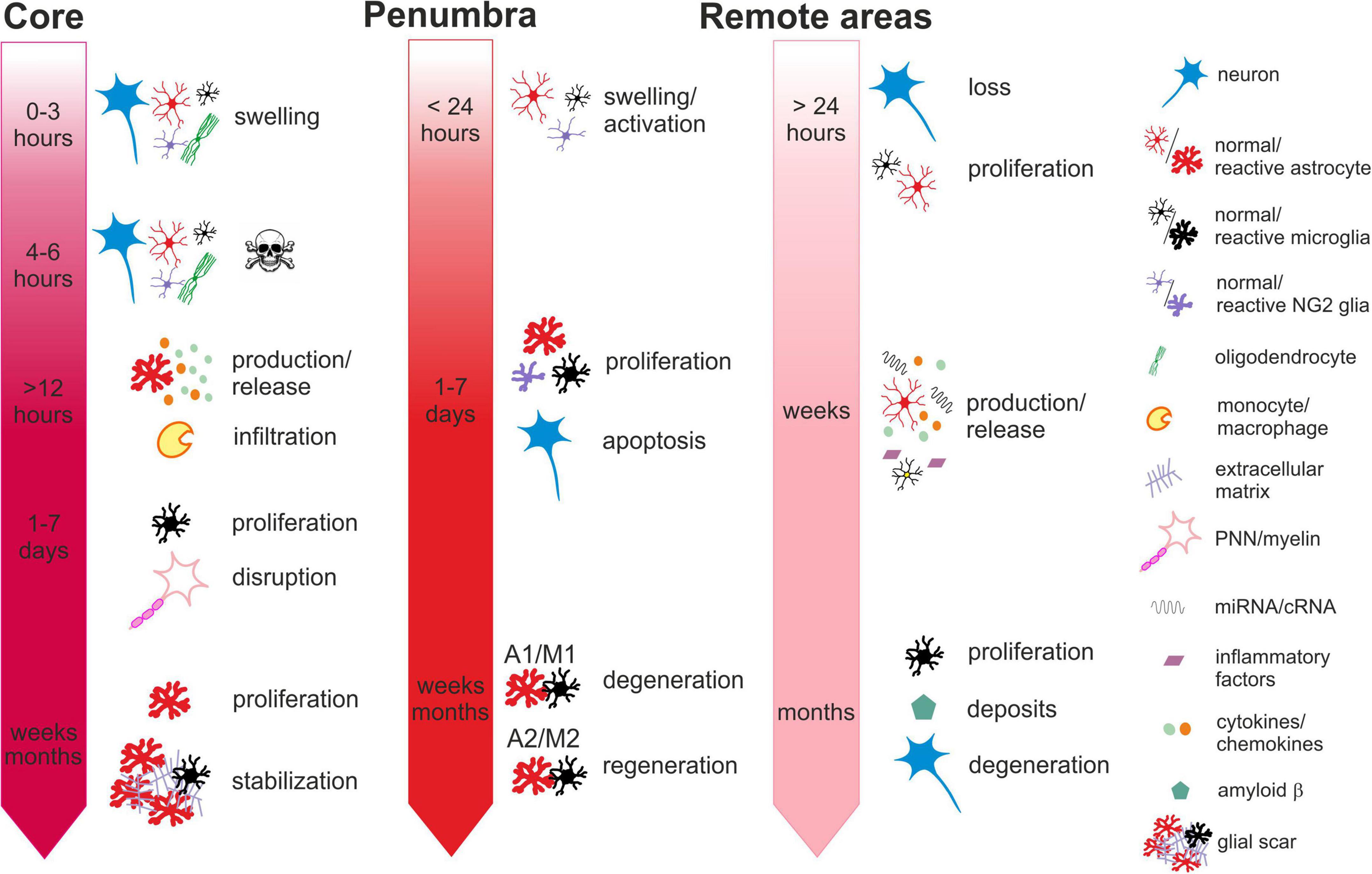
Ischemia-induced changes in the core, penumbra and remote areas in time are described in more details in the chapters below and summarized in Figure 1.
Figure 1. Time-dependent ischemia-induced changes in the ischemic core (left), penumbra (middle) and remote areas (right). Core: Profound cell swelling occurs several minutes after the onset of ischemia. Within 6 hours, neurons undergo necrosis and general loss of cells can be observed. Around 12 hours after ischemia, the surviving astrocytes begin to release chemokines attracting migration and infiltration of monocytes/macrophages. One to seven days after ischemia, the number of cells increases mostly due to the infiltration of microglia and macrophages and the massive proliferation of microglia. The numbers of neurons are severely reduced, and perineuronal nets and myelin sheaths are disrupted. One month or more after ischemia, the number of astrocytes increases, due to their proliferation and migration from the penumbra. The lesion contracts and the glial scar is stabilized by the extracellular matrix produced by reactive astrocytes. Penumbra: Swelling and activation of astrocytes, NG2 glia and microglia are delayed in comparison with the core but occur within 24 hours after ischemic insult. One to seven days after ischemia, neurons are still visible but their numbers have declined due to apoptosis. Astrocytes, microglia, NG2-glia and oligodendrocytes intensively proliferate. Proliferating astrocytes and microglia create heterogenic groups, where distinct subtypes differ in gene expression and membrane properties. Astrocytes and microglia with pro-inflammatory phenotypes in the vicinity of the core contribute to the formation of glial scar. Those with anti-inflammatory phenotypes in the outer parts of the penumbra with less severe hypoperfusion begin to produce growth factors and cytokines contributing to tissue regeneration. One month or more after ischemia, the numbers of glial cells are moderated, and depending on the duration and severity of the hypoperfusion, neurodegenerative or regenerative processes are activated. Remote areas: The first changes in the remote areas can be observed not earlier than 24 hours after ischemic insult but typically several days, weeks or even months following it. Depending on the brain region, changes in the cell structure and numbers are subtle or there are none. A slight decline in the numbers of neurons and a moderate increase in the numbers of microglia or astrocytes can be observed. However, morphological changes typical for their reactive states are mostly missing, even though the cells express markers of activation. More distinct alterations can be observed in the cellular functions, gene and protein expression profiles or production of cytokines. Induced delayed neuroinflammation may evoke neurodegenerative processes and amyloid deposits. For more details see the text.
3.1 Regional variability in zone definitions in rodent models of focal ischemiaThe common regions of the necrotic core in rodent models of focal ischemia (see Glossary) are well known. Phototrombotic model of MCA blockage causes reproducible infarcts involving the parietal cortex in all its layers (Dihne et al., 2002; Reichmann et al., 2002; Schroeter et al., 2002; Haupt et al., 2007; Karetko-Sysa et al., 2011); other special types of phototrombotic models may cause necrosis for example in the caudoputamen (Kuroiwa et al., 2009). The striatum and cortex are consistently affected by the MCA occlusion (MCAO) in its proximal section using the Longa method (Tanaka et al., 2001; van Groen et al., 2005; Melani et al., 2006; Justicia et al., 2008; Bona et al., 2019) although the size may vary (Arlicot et al., 2010). On the other hand, the outcome of the proximal MCAO according to Koizumi, seems to be dependent on the duration of blood flow cessation. Short-term MCAO lasting 30 min leads to necrosis bound to the striatum (Arvidsson et al., 2001; Kronenberg et al., 2012; Prinz et al., 2015) longer occlusions (45–120 min) begin to involve the parietal cortex (Arvidsson et al., 2001; Bacigaluppi et al., 2009; Garbuzova-Davis et al., 2014; Cai et al., 2017; Gaire et al., 2019), and a 3-h occlusion spreads even further to the globus pallidus (Dihne and Block, 2001). The permanent version of the Koizumi method creates infarctions spanning over the striatum and almost the entire ipsilateral cortex (Takasawa K. et al., 2002; Ni et al., 2020). Distal MCAO produces reliable infarcts restricted to the somatosensory cortex (Hobohm et al., 2005; Ling et al., 2009; Cao et al., 2021; Ip et al., 2021). Researchers tend to get very similar results concerning the area of the ischemic core, yet for example, Popp et al. (2009) assume, that the core created after 120 min of proximal MCAO, also involves the amygdala and hypothalamus on top of the striatum and cortex, and that the penumbra spans into the hippocampus, thalamus and part of the hypothalamus (Popp et al., 2009). The thalamus, which has been extensively studied for its remote effects is, in the vast majority of publications, described as a primarily nonaffected structure (Dihne et al., 2002; Justicia et al., 2008), just like the hippocampus (Uchida et al., 2010). The discrepancies in the assignation of specific brain regions to either ischemic core, penumbra or remote areas may stem from the various methods used for their definitions. Thus, some studies rely on the definition depending on the acute reductions of blood flow (Sakoh et al., 2001; Yu et al., 2016), changes in the regional glucose metabolism (Szilagyi et al., 2012), the combination of hypoperfusion and the damage to the dendritic structure (Li and Murphy, 2008) or deviant signal intensity via MRI (Buffon et al., 2005).
4 The time course of stroke-induced changes in the core and the penumbra 4.1 Acute and sub-acute post-stroke reactionThe severely hypoperfused core is the location of the first reaction to ischemia. The first tissue reaction to the oxygen deficit is a substantial decrease in number of NG2-glia (Lee et al., 2003) and swelling of oligodendrocytes (Hernandez et al., 2021). Within the first 3 h post-ischemia, neurons with hyperchromatic nuclei can be observed (Yang et al., 2013). Four to 6 h following ischemia, the endothelial cells of the local capillaries become activated and the compromised integrity of the BBB results in edema. Hematoxylin-eosin staining can reveal red neurons indicating their damage (Hirouchi et al., 2007) and a general loss of cells. Barely any astrocytes can be seen (Nowicka et al., 2008; Aleithe et al., 2019) and those that remain are the source of the monocyte chemoattractant protein-1 (MCP-1) at 12 h after ischemia (Che et al., 2001). One day post-ischemia, macrophages/microglia are recruited into the core and their somas become larger, resembling an amoeboid shape. The tissue loses its clear structure, there is almost no immunostaining signal for neurons and the loss of Wisteria floribunda (WFA) staining with preserved proteoglycans indicates the partial decomposition of perineuronal nets (PNNs) (Hobohm et al., 2005). Axons begin to disappear (Khodanovich et al., 2018). The activated microglia and astrocytes also appear in the penumbra, and the astrocytes begin to form a wide layer around the ischemic core (Mabuchi et al., 2000; Melani et al., 2006; Nowicka et al., 2008). Glia are one of the main secretors of the ECM deposited in the lesion and the accumulated ECM macromolecules prevent axonal outgrowth in the area void of neurons (Dzyubenko et al., 2018). Both the NG2-glia (Tanaka et al., 2001) and astrocytes (Melani et al., 2006) are swollen, but the cell numbers do not differ. Although the number of neurons in the penumbra declines (Melani et al., 2006), the staining intensity for neuronal nuclear protein (NeuN) and microtubule-associated protein (MAP2), markers of mature neurons, remains constant (Cao et al., 2021). In contrast to the core, the PNNs in the penumbra are intact.
4.2 Early chronic post-stroke reactionMassive gliosis spreads in the core on the third day after ischemia, with microglia proliferating to a large extent (Che et al., 2001); an increase in the number of glial cells can also be observed in the penumbra (Gaire et al., 2019). The release of the MCP-1 for the attraction of immune cells is intensified in the core and its spreading into the surroundings can also be observed (Che et al., 2001). Myelin sheaths in the core are damaged, the affected axons inside them separate from each other and vacuolization can be detected (Khodanovich et al., 2018). In the penumbra, neurons are still visible, in contrast to the core (Cao et al., 2021) and neural stem cells (NSCs) begin to appear in both structures (Shin et al., 2013). The MCP-1 expression dissappears from the core 5 days following ischemia, but still dwells in its outer rim (Che et al., 2001). One week after focal ischemia, the core still displays a minimum of neuronal cells and necrotic debris occupies the void (Schroeter et al., 2002). The phagocytic activity peaks at this time (Toth et al., 2016) as the core is packed with microglia (Michalski et al., 2017). NG2-glia are absent in the core, unlike in the penumbra, where they grow in size and numbers (Tanaka et al., 2001), presumably due to migration from the subventricular zone (SVZ) (Hernandez et al., 2021). Some of them acquire astrocyte-like phenotype and contribute to the formation of the glial scar (Valny et al., 2018). The structure overflows with astrocytes, microglia and oligodendrocytes (Mabuchi et al., 2000), and the glial scar becomes thinner as the astrocytes align more closely to each other (Hobohm et al., 2005; Nowicka et al., 2008; Cao et al., 2021). Ischemic injury evokes a reduction of NeuN+ neurons and a complete loss of MAP2+ neurons in the infarct core. In peri-infarct areas, the number of NeuN+ neurons is also decreased, but MAP2+ neurons are located around the infarct border (Cao et al., 2021). Hobohm et al. (2005) observed an appearance of aggrecan expression in reactive astrocytes 7 days after MCAO; the glial scar is already clearly visible at this time (Hobohm et al., 2005). The intensity of microglial Iba-1 staining reaches its peak in the core 2 weeks post-ischemia (Reitmeir et al., 2011). Myelination of the tissue remains low (Khodanovich et al., 2018) with a decline observed in the penumbra (Tanaka et al., 2016).
4.3 Delayed chronic post-stroke reactionOne month after ischemia, some axons are entirely demyelinated which negatively affects the interneuronal transport (Garbuzova-Davis et al., 2014). The loss of neurons is even more profound (Cao et al., 2021). The lesion contracts and contains a lot of swelling astrocytes, whose endfeet at the tip of the degenerating processes are detached from the capillary within the neurovascular unit (Nowicka et al., 2008). Microglia can be observed phagocytising around blood vessels (Garbuzova-Davis et al., 2014). The core is surrounded by a layer of polarized astrocytes with their processes extended toward the lesion (Nowicka et al., 2008). The immense numbers of glia in the penumbra are now moderated (van Groen et al., 2005). It takes a few more weeks for reactive microglia to diminish in the core, while the astrocytic scar becomes even more distinct (Reitmeir et al., 2011; Cao et al., 2021). Even 4 months after ischemia, some degenerated neurons may still be seen in the core and the total number of neurons is substantially attenuated (Cao et al., 2021).
5 The post-ischemic response in specific structures of the remote areasFocal ischemic injury does not impact all remote regions in the same way. There may be various explanations for this heterogeneity, for instance, the existence of anatomical connections between the areas of the primary and the secondary lesion (Chen et al., 2014), unique vasculature (El Amki et al., 2015), the high density of receptors that mediate damage or recovery (Arvidsson et al., 2001; Onufriev et al., 2021).
5.1 Ipsilateral cortexThe cortex is typically the site of the ischemic core in the focal ischemia. However, only a part of the cortex can be truly necrotic, the rest may seem intact at first glance, especially during the acute phase. Apoptosis of neurons does not seem to occur in the remote cortex (Karetko-Sysa et al., 2011), yet this does not mean that cells are not impacted. For example, some researchers observed neurodegeneration in the remote ipsilateral cortex (Chen et al., 2014; Bona et al., 2019), although others claim no such damage was seen in their samples (Melani et al., 2006; Karetko-Sysa et al., 2011). It can be hypothesized that these different results might be attributed to the different time of post-stroke tissue evaluation (Minassian et al., 2019) or the type of an ischemic model (permanent versus reversible MCAO), where reperfusion can lead to more severe injury by the rapid burst release of reactive oxygen species (ROS) (Peters, 2006). Other neuron-associated changes, such as degeneration of axial dendrites with vacuolization and partial loss of synapses, were observed 7 days after photothrombosis, with a progressive deterioration in the following weeks (Lee et al., 2020). The increase in protein levels of growth associated protein-43 (GAP-43), a marker of axonal growth cones, indicates parallel damage and regeneration in the peri-ischemic cortex (Chen et al., 2014).
It was detected that the quantity and morphology of astrocytes in the ipsilateral medial frontal and cingulate cortex were constant 24 h after permanent MCAO (pMCAO) (Melani et al., 2006). Later stages are characterized by an elevation in GFAP expression, especially 4 and 7 days after ischemia, implying hypertrophy and increased proliferation of astrocytes (Nowicka et al., 2008). Ischemia ignites a wave of spreading depression in the astrocytic syncytium and the effects can spread into remote areas (Haupt et al., 2007). The number of cells expressing mRNA of connexin 43 (Cx43), the main component of astrocytic gap junctions (Liang et al., 2020), was reduced on day 1 following ischemia, slightly elevated on day 3, and substantially increased on day 7 in comparison to the contralateral (control) cortex. Subsequently, the amount of Cx43 mRNA-positive cells returned to levels comparable to the control 2 weeks after the ischemic injury (Haupt et al., 2007). These results indicate the temporary alterations in the intercellular connections within the astrocytic syncytium, that may affect the spread of calcium waves and injury propagation.
The remote cortex is a location where an inflammatory response can also be triggered, as was confirmed by the presence of heat shock proteins (Popp et al., 2009) and microglia. Microglial numbers were found to be highly elevated 1 and 3 days after proximal transient MCAO (tMCAO) (Gaire et al., 2019), just like in the core and penumbra, nevertheless, a closer look at the morphomolecular cell characteristics may show distinct reactions in each zone. It was proposed, that an intermediate state between resting and fully activated microglia exists - these cells have thin ramified processes similar to resting microglia, yet they expressed a marker of microglia activation, the purinergic receptor P2X7 (Melani et al., 2006; Monif et al., 2009).
In contrast to microglia, the NG2-glia seem to remain quiescent during ischemia in the remote areas, while they swell in the penumbra (Tanaka et al., 2001). To the best of our knowledge, no studies have investigated oligodendrocytes in the cortex beyond the penumbra so far.
The cortical ECM partially disappears upon ischemia. WFA staining revealed a decreased density of PNNs already 4 h after MCAO with an even more pronounced decline occurring 24 h after ischemia and restoration in the following week. The brevican immunoreactivity was also transiently attenuated 1 day after ischemia. This decline in ECM levels may be explained by enzymatic digestion (overproduction of MMPs and/or hyaluronidase) or reduced production of ECM by inhibitory neurons. On the other hand, ischemia did not affect the expression of aggrecan (Karetko-Sysa et al., 2011) or neurocan (Deguchi et al., 2005). One of the explanations for this diversity may be the contradicting actions of activated astrocytes and microglia, which produce both molecules of the ECM and the enzymes responsible for its degradation. Moreover, microglia may remove the macromolecules from the ECS by phagocytation (Dzyubenko et al., 2018; Raffaele and Fumagalli, 2022).
5.2 HippocampusThe hippocampus is traditionally regarded as highly susceptible to global ischemia (Nikonenko et al., 2009; Baron et al., 2014), however, even focal ischemic injury can have a profound impact on it. The hippocampal damage in the ipsilateral hemisphere after focal ischemia is manifested, for instance, by the massive increase in apoptotic cells 2 weeks after tMCAO (Yang et al., 2019).
The first signs of neuronal degeneration can already be seen 12 days after MCAO in the cornu ammonis (CA) (Butler et al., 2002). Significantly reduced numbers of neurons can already be detected after 3 days (Uchida et al., 2010) and persist as long as 12 months after ischemia (Ouyang et al., 2020). A study by Park et al. (2011) was focused on the specific subsets of neurons in the CA1 and CA3 regions. They observed a decline in the numbers of cholinergic, NO-positive (NO+) and nitric oxide synthase-positive (NOS+) neurons, which were associated with impaired learning and memory in experimental animals (Park et al., 2011). In contrast, the study of Uchida et al. (2010) revealed increased levels of neuronal and inducible NOS (Uchida et al., 2010). Furthermore, they detected increased amounts of superoxide dismutase, which indicates augmented antioxidant activity.
However, other studies did not find any changes in neuronal numbers in the hippocampus, possibly due to only a brief interruption of the oxygen supply and examination of the histological results after 1 month, when the tissue might have undergone partial regeneration (Zhou et al., 2013; Brait et al., 2021). To compensate for the neuronal loss, the expression of doublecortin-expressing stem cells is increased in the subgranular zone (SGZ) of the dentate gyrus (Klein et al., 2016), which is one of the regions with preserved neurogenesis in adult life. The number of proliferating cells increases on the first day after proximal pMCAO, and reach their peak 4 days after ischemia. Around half of the progenitor cells begin to differentiate into neurons, whereas the other half develop into astrocytes (Takasawa K. et al., 2002).
The reaction of astrocytes to remote ischemia in the hippocampus are consistent: the GFAP levels and the numbers of GFAP-positive (GFAP+) cells increase shortly after the stroke (Haupt et al., 2007; Nowicka et al., 2008; Uchida et al., 2010) and remain elevated for several months (Ouyang et al., 2020; Brait et al., 2021). Butler et al. (2002) linked the augmentation of astrocyte activation with the appearance of degenerating pyramidal neurons (Butler et al., 2002), which likely occurred as a result of the astrocytic release of neurotoxic molecules (Phatnani and Maniatis, 2015). The time course of Cx43-positive cell numbers in the CA1 region differs from the one in the cortex: the boost starts from the first day and the rise is even stronger 1 week after ischemia (Haupt et al., 2007).
Experiments with proximal MCAO in male rodents led to substantial increases in Iba-1 protein levels and numbers of Iba-1+microglia (Uchida et al., 2010; Brait et al., 2021). However, MCA photothrombosis in female mice did not impact the hippocampus, as this structure was only surrounded by activated microglia (Schroeter et al., 2002). These varying results may be attributed to the use of animals of different sexes as female mice are less susceptible to macrophage infiltration than males (Xiong et al., 2015). Alternatively, the use of the photothrombotic method, which creates precisely defined trauma may have prevented excessive spreading of the injury to the hippocampus (Clark et al., 2019).
Uchida et al. (2010) examined the fate of oligodendrocytes after ischemia and found a gradual decrease in their numbers with significant alterations in the third and seventh day after ischemia (Uchida et al., 2010).
Studies regarding the hippocampal ECM detected decomposed PNNs (Hartig et al., 2017) and attenuated immunostaining for type IV collagen, which could be explained by the vast expression of MMP-9 (Yang et al., 2019). The volume of the whole structure remained stable for the first 12 weeks after ischemia, followed by a minor yet significant enlargement (Brait et al., 2021).
5.3 ThalamusThe thalamus is the most explored structure in the subject of secondary post-ischemic damage. The intense focus on this region may be due to the existence of the physical axonal connection with the primary site of the insult, which can explain the diaschisis (Ouyang et al., 2020). There is already vast evidence showing that the thalamus is indeed impacted by ischemia in distant regions.
Hartig et al. (2016) found shrunken and fragmented GABAergic neurons from 1 day after ischemia in the reticular thalamic nucleus (Hartig et al., 2016). In contrast, Loos et al. (2003) did not see any abnormal morphology nor any decline in the number of neurons at that time (Loos et al., 2003). The discrepancy may be explained by the longer exposure to ischemia (pMCAO vs. proximal tMCAO) in the case of Hartig’s study or the use of different markers for neurons; parvalbumin expression can be found only in a subset of nerve cells, whereas NeuN is considered a pan-neuronal marker (Gusel’nikova and Korzhevskiy, 2015; Hartig et al., 2016). Most of the studies report the first changes occurring in the thalamus 1 week after focal ischemia, showing aberrant intercellular content (Dihne et al., 2002) and neuronal loss irrespective of the animal model used (Hirouchi et al., 2007; Wang et al., 2007; Ling et al., 2009; Chen et al., 2014; Ladwig et al., 2019; Xu W. et al., 2020). The levels of autophagy-related proteins Beclin 1 and MAP1LC3 (microtubule-associated protein 1A/1B-light chain 3) significantly rise (Xu W. et al., 2020). However, the number of neuronal cells remains reduced for months (Justicia et al., 2008).
Unlike in the ischemic core, astrocytes in the thalamus do not undergo any changes the first day after ischemia (Loos et al., 2003). The first signs of astrogliosis may be detected the third day after MCAO, which evolves fully 1 or 2 weeks after ischemia (Loos et al., 2003; Cao et al., 2021). The changes in activated astrocytes included upregulated GFAP expression (Loos et al., 2003; Hobohm et al., 2005), increased numbers (Dihne et al., 2002; Hirouchi et al., 2007; Xu W. et al., 2020) and cell swelling (Loos et al., 2003). Moreover, several studies even reported the formation of astrocytic scar (van Groen et al., 2005; Justicia et al., 2008). In contrast, one research group did not find any alterations in GFAP expression from 4 to 60 days after ischemia (Nowicka et al., 2008). Interestingly, Ling et al. (2009) reported the appearance of NSCs with morphological features of astrocytes, which grow in numbers the second week after ischemia and stretch first along the WM fibers and later accumulate in the ventroposterior thalamic nucleus (VPN) (Ling et al., 2009).
Similarly to the cortex and hippocampus, it is possible to observe activated microglia in the thalamus around the seventh day after ischemia, first with the hyper-ramified and later swollen shape (Dihne et al., 2002; Schroeter et al., 2002; Hobohm et al., 2005; Hirouchi et al., 2007; Wang et al., 2007; Ling et al., 2009; Klein et al., 2016; Ladwig et al., 2019; Xu W. et al., 2020). Of note, microglia-related genes are strongly upregulated in the ipsilateral thalamus a few days before the cell activation and the microgliosis persists for several months (Justicia et al., 2008; Cao et al., 2021). Chronic microglia activation is associated with the development of neurodegenerative diseases and can be an important link between stroke and post-ischemic Alzheimer’s or Parkinson’s disease (Lull and Block, 2010). Additionally, Smirkin et al. (2010) identified a subpopulation of microglia staining positively for Iba-1 and NG2, which is a distinct glia lineage with possibly neuroprotective properties (Smirkin et al., 2010). Interestingly, Ladwig et al. (2019) noticed an inverse proportion between the number of neurons and microglia: the more microglia, the fewer neurons (Ladwig et al., 2019). It was also observed that as soon as activated microglia appear in the thalamic tissue, the neurons begin to degenerate (Cao et al., 2021). Microglia were observed concentrated around amyloid β precursor protein (APP) deposits (Justicia et al., 2008). APP deposits, as well as amyloid β (Aβ) and Aβ plaques, can appear in the thalamus within weeks after ischemic insult and can persist for months (van Groen et al., 2005; Wang et al., 2007; Justicia et al., 2008; Lipsanen et al., 2011). The microglia can also collect iron, which may be accumulated in the APP deposits (Justicia et al., 2008). Moreover, the same study revealed increased transcription of heme oxygenase-1 (HO-1), an indicator of oxidative stress. All the processes imply the development of the post-ischemic tissue toward neurodegenerative dementia (Justicia et al., 2008). However, it is not possible to claim that solely microglia trigger neurodegeneration, although they substantially contribute to this process [for review, see (Harry, 2021)]. Clinical studies on post-ischemic secondary injury are rare, therefore we cannot draw any conclusions from them yet.
Another noteworthy event in the thalamus is the damage of the ECM molecules. WFA staining is strongly reduced from the PNNs the first day after ischemia; this effect is even more pronounced in aged mice (Hartig et al., 2016). The expression of other constituents of PNNs, such as aggrecan and neurocan, and generally of the chondroitin sulfate proteoglycans (CSPGs), is downregulated (Hobohm et al., 2005; Hartig et al., 2016).
Degenerating tracts of white matter between the thalamus and the cortex and atrophy of the entire structure were detected after stroke (Reichmann et al., 2002; Arlicot et al., 2010). In contrast to the ipsilateral cortex, the thalamic levels of GAP-43 and synapsin (the marker of synaptic vesicles) were diminished (Wang et al., 2007; Chen et al., 2014). This may suggest that synaptic regeneration is delayed in the thalamus. Alternatively, there are homeostatic control mechanisms counteracting the pathological direction of the thalamic tissue. For instance, cell proliferation begins no later than 1 week after ischemia and the intensity further increases in the second week (Ling et al., 2009). Some nestin and GFAP+ cells were detected along the corticothalamic fibers and in the VPN. These cells can later differentiate either into astrocytes or neurons (Witusik et al., 2008).
Tissue regeneration may be manifested in angiogenesis, i.e., the growth of new blood vessels importing oxygen and nutrients (Hatakeyama et al., 2020). Ischemia has been found to increase vascular density (Yanev et al., 2017), and capillary thickness and to trigger the proliferation of endothelial cells in the ipsilateral thalamus (Ling et al., 2009). The process is presumably stimulated by the release of angiogenic cytokines such as VEGF, MMPs, angiopoietins or basic fibroblast growth factor (BFGF) from the penumbral cells (Fang et al., 2023). Consequently, the volume of the thalamic blood flow increases (Yanev et al., 2017).
5.4 Substantia nigraAlthough the substantia nigra (SN) is situated farther away from the common areas of the ischemic core, the secondary damage is evident. This might be caused by the disturbed interregional fiber connections (Hirouchi et al., 2007; Prinz et al., 2015), such as the nigrostriatal tract (Sonne et al., 2023). SN is the most explored brainstem structure for its crucial role in motor function, which frequently deteriorates after stroke (Prinz et al., 2015).
Neurons in SN are the first type of cells to respond to remote ischemia. The use of an electron microscope allowed for the observation condensed neuronal chromatin on the first day and degraded endoplasmic reticulum the second day after ischemia (Zhao et al., 2002). These details were not detected by other research groups, which reported healthy-looking neurons during the first week (Dihne and Block, 2001; Loos et al., 2003). Four days after ischemic insult, vacuolation of neurons occurs and their numbers start to decline. In cells with condensed cytoplasm, the plasmalemma later disintegrates (Zhao et al., 2002) and phagocytes remove the dead neurons (Dihne and Block, 2001). The cell loss is extensive, with a 52% decrease in neuronal numbers observed. This results in atrophy of the entire structure that can be detected 7 days after ischemia, and even more distinct shrinkage was evident 2 weeks following MCAO (Dihne and Block, 2001). In addition, the number of dopaminergic neurons, which comprise an important subpopulation of neurons in the SN, were also found to diminish the first week after ischemia (Huh et al., 2003; Prinz et al., 2015) and this decline was confirmed even several months following MCAO (Kronenberg et al., 2012). Nerve cells may transiently participate in the production of anti- or pro-inflammatory cytokines (Doll et al., 2014). In the remote SN, the intracellular presence of TNF-α (Loos et al., 2003) and IL-6 (Dihne and Block, 2001) were detected.
The microglia in SN become activated within the first week and their numbers increase (Prinz et al., 2015). Their shape becomes more hypertrophic, but transition into an ameboid shape was never observed (Dihne and Block, 2001; Huh et al., 2003). Similarly to the cortex, microglia in the SN are transformed into an intermediate semi-activated state. The duration of microglia activation seems to be somewhat shorter than in other regions as a maximum of 2 months was reported (Huh et al., 2003).
The changes observed in microglia are essentially very similar to those of the astrocytes. Astrocytes gradually swell (Loos et al., 2003), proliferate (Hirouchi et al., 2007) and their GFAP expression is upregulated (Dihne and Block, 2001), yet the intracellular space appears non-aberrant (Zhao et al., 2002).
In contrast, oligodendrocytes stay intact for several weeks after the insult (Zhao et al., 2002). Nevertheless, the effect of ischemia is potent enough to trigger angiogenesis and increase the perfusion of the SN (Yanev et al., 2017). No reports addressing alterations of the ECM in this region have yet been published.
5.5 White matterThe state of WM can be reflected by the post-stroke behavioral outcome as was reported by a clinical study, where a correlation between a long-term cognitive decline and a low integrity of exo-focal WM was observed (Schaapsmeerders et al., 2016). A comparable situation was seen in an animal study, where proximal tMCAO in aged mice caused severe demyelination of the corpus callosum (CC) 8 weeks after ischemia, which was associated with poor results in the corner and cylinder tests (Cai et al., 2017). Even more detailed results were shown by Wan et al. (2022), who reported that distal MCAO in young mice led to a significant decrease in the levels of myelin basic protein (MBP), myelin-associated glycoprotein (MAG), and neurofilament 200 (NF200) in the CC 7 days after ischemic insult. In addition, myelin density was diminished, the myelin integrity weakened, and its thickness was reduced by approximately one-half; the percentage of myelinated axons attenuated, and the diameter of myelin sheaths was reduced. The authors suggested that astrocytes may be partially responsible for the demyelination by their phagocytosis of ischemia-damaged myelin debris. They detected an increase in levels of GFAP protein and a reduction in the amount of an anti-inflammatory marker S100A10 (S100 calcium-binding protein A10). Furthermore, they detected an augmentation in levels of pro-inflammatory protein C3d (Complement component 3d), and an increased amount of lipocalin-2 protein, which is released by activated astrocytes and may be responsible for myelin degradation. All these astrocyte-related changes occurred at the same time as the WM destruction (Wan et al., 2022). However, not all animal studies concluded that ischemia must necessarily harm the WM. For example, one study found that the density of myelin fibers in the CC was unaffected 4 weeks after transient ischemia (Zhou et al., 2013). Reitmeir et al. (2011) obtained similar results in corticospinal tract fibers 52 days after ischemia (Reitmeir et al., 2011). Similarly, another study focusing on exo-focal changes in the thalamus and examining the internal capsule between the ischemic cortex and the thalamus did not find any pathological abnormalities (Dihne et al., 2002). It is difficult to establish a pattern which would explain these contradictory results. One may argue that senescence is a great contributor, as the extent of WM injury was found to be age-dependent (Rosenzweig and Carmichael, 2013). For example, one retrospective study (Yu S. et al., 2018), conducted on children with a history of perinatal stroke, revealed significantly lower myelinization in the contralateral hemispheres when compared to healthy controls, and even more profound loss of myelin in the ipsilateral remote areas. Other determinants may be the experimental species and the type of focal ischemia model.
As for cellular appearance in the fiber tracts, nestin-positive cells with astrocytic phenotype were detected in subcortical fiber tracts the first week after distal MCAO, before they spread into the adjacent thalamus (Ling et al., 2009). Interestingly, ischemic injury evokes the upregulation of hyaluronan (HA) throughout the ipsilateral CC and the peri-infarct area and accumulation of HA within the glial scar surrounding the lesion. Ischemia also induces the upregulation of HA synthase 2 (HAS-2) and hyaluronidase Hyal2 production, as well as the expression of Rhamm (hyaluronan receptor) in astrocytes (Lindwall et al., 2013). Greda and Nowicka (2021) investigated HA metabolism in the ischemic brain and found that all 3 synthases (HAS1-3) and 2 hyaluronidases (Hyal1, 2) were affected. The authors also showed that inhibition of hyaluronidase improves behavioral outcomes after stroke (Greda and Nowicka, 2021). Additionally, cell culture experiments suggest that parameters, such a
留言 (0)