

記住我



Neurodegenerative disorders are a collection of disorders that manifest as a slow degeneration of nerve cells and are particularly common in the elderly. With their insidious onset and irreversible progression, human health is greatly threatened by neurodegenerative diseases. The world population is continuing to age, and as a result, mortality and morbidity rates related to neurodegenerative disorders are rising, significantly raising the socio-economic burden (Zhang et al., 2021). However, these diseases can only be alleviated, and it is difficult to make a breakthrough in their treatment (Heemels, 2016).
Targeting RAS, eratin is a tiny molecule that is selectively deadly and was discovered in tumor cells in 2003. This chemical was shown to induce a distinct kind of non-apoptotic cell destruction (Dolma et al., 2003). An innovative type of iron-dependent cell death termed ferroptosis, which is induced by erastin, was identified in 2012 by Stockwell's laboratory. Ferroptosis differs from autophagy, necrosis, and other types of cellular destruction due to inherited, biochemical, and structural variations (Dixon et al., 2012). The basic ferroptosis process is the Fenton reaction, which is driven by ferrous ions and is responsible for initiating the peroxidation of lipids on the cell surface. This process is defined by diminished mitochondrial cristae, rising membrane density, and atrophy of the mitochondria (Vitalakumar et al., 2021). Considering its particular role in numerous clinical conditions, including brain diseases, tumors, and tissue damage resulting from ischemia-reperfusion, it has garnered significant attention (Conrad et al., 2016; Stockwell, 2022). A substantial body of evidence indicates that ferroptosis contributes to the pathophysiology of neurological disorders, and is associated with peroxidation of lipids, brain iron accumulation, and glutathione deficiency. Such conditions have the potential to trigger a series of events that include neurotransmitter oxidative stress, myelin deterioration, inflammatory activation, loss of neuronal interaction, memory loss, and cellular death. Nevertheless, the mechanism that triggers cell death is unknown. Thus, more research is needed to understand how pathologic traits, pathogenesis, and ferroptosis interact in neurodegenerative diseases from a therapeutic perspective.
2 Ferroptosis's relation to other cellular death programsThe Nomenclature Committee on Cell Death divided controlled apoptosis into multiple categories in 2018: pyroptosis, autophagy, ferroptosis, necrosis, and apoptosis. This classification was based on the structural, biochemical, and therapeutic perspectives (Galluzzi et al., 2018). In vivo, cell membrane breakdown from oxidative damage causes an overabundance of reactive oxygen species (ROS) that causes lipids to peroxide, which leads to multiple kinds of cellular death (Su et al., 2019). The peroxidation of lipid products interacts with inhibitors and transcription variables, membrane receptors, and initiates apoptotic transmission (Elkin et al., 2018). Since ferroptosis occurs later in membrane rupture, phosphatidylserine ectopics may occur, which is similar to apoptosis (Tong et al., 2022). As a defense mechanism, autophagy divides misfolded proteins and damaged organelles using a dual membrane structure named an autophagosome, then integrates them with lysosomal phagocytosis of contents (Sevenich, 2018). Thus, to preserve equilibrium and regular activity of cells in vivo, the autophagic lysosomal pathway is significant. The ferroptosis inducer erastin induces autophagy, and a significant moderator of ferritin destruction and transferrin receptor 1 (TfR1) expression is reactive oxygen species (ROS)-induced autophagy (Park and Chung, 2019). Nuclear receptor coactivator 4 (NCOA4) was shown in research to activate ferritin phagocytosis and heat-shock protein 90 (HSP90)-associated chaperonin-mediated autophagy to promote ferroptosis (Zhou et al., 2020). An inflammatory response can be triggered by pyroptosis, a pro-inflammatory kind of programmable cell death (Chang et al., 2020), and exhibits an acute disruption of the cell membrane and an intracellular secretion of pro-inflammatory molecules, involving interleukin (IL) 18 and IL-1β (Man et al., 2017). It was found that CD8+ T lymphocytes, known as antitumor immune cells, may induce and increase ferroptosis and pyroptosis (Tang et al., 2020). Firstly, CD8+ T-cells may release human granzyme family members (GZMA proteins) to act as cleaving enzymes for Gasdermin- b, Gasdermin structural domain protein family member (GSDMB). Cleavage by GSDMB leads to pyroptosis. However, the Solute carrier family 7 (SLC7A11) member is reduced as a result of CD8+ T cells releasing IFN-γ, causing lipid ROS accumulation and suppressing ferroptosis. Therefore, the combined study of ferroptosis and Pyroptosis promotes in development of cancer therapy. Research showed that cellular damage, such as ferroptosis and pyroptosis, might intensify the process of tau hyperphosphorylation and an inflammatory response linked to AD progression (Qiu et al., 2023). Thus, different cell death processes may interact with each other and be closely linked to disease progression.
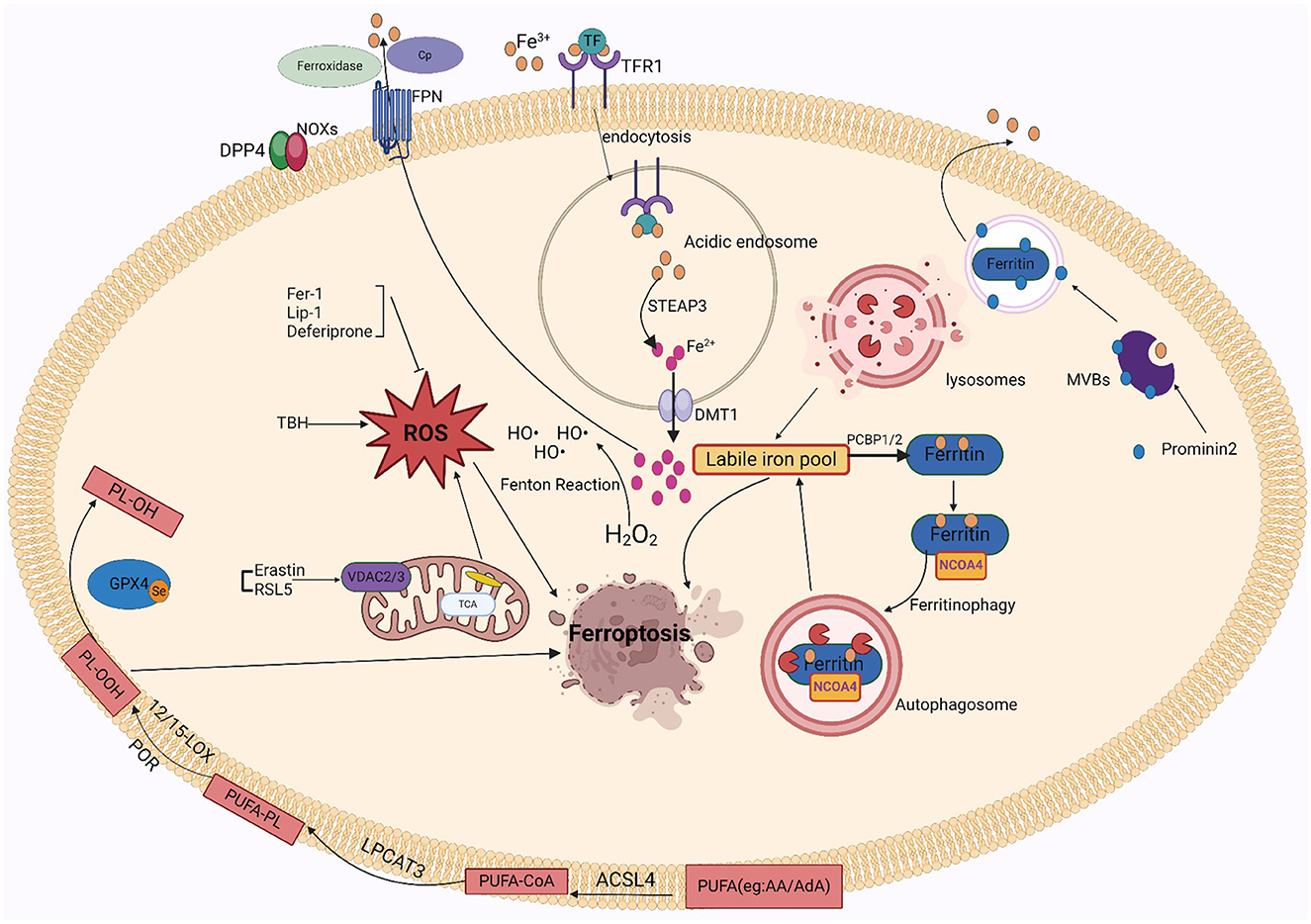
3 Mechanisms of ferroptosisFerroptosis is characterized by unchecked peroxidation of lipids and consequent disruption to the cellular membrane, its onset and development are controlled by intricate signaling and regulatory mechanisms. Susceptibility to ferroptosis depends on iron accumulation, glutathione (GSH) depletion, antioxidant inactivation, polyunsaturated fatty acids (PUFAs), and selenium availability (Jiang et al., 2015; Doll et al., 2017). These proteins or metals are important in regulating the degree of oxidative stress within cells. Iron is a significant trace element involved in multiple physiological processes, such as metabolizing energy, transport of oxygen, and oxygen preservation. The internal balance of iron is carefully controlled by the body. Since iron cannot be removed from the body through clearance mechanisms, its regulation in the body is critical. Irreversible oxidative damage occurs when iron is overloaded and exceeds its storage capacity. Several ROS sources that can trigger ferroptosis include the iron-mediated Fenton reaction, mitochondrial ROS, and membrane-associated ROS carried on by the nitrogen oxides (NOX) family of proteins. Pathological states, in which ROS levels are drastically elevated, can cause severe damage to cellular structures. Ferroptosis is directly mediated by lipid peroxidation. Phospholipids comprising polyunsaturated fatty acids, specifically arachidonic acid (AA) or adrenoic acid (AdA) within the phosphatidylethanolamine (PE) molecule, are the primary peroxidation of lipid precursors in iron overload (Galaris et al., 2019; Zhong et al., 2019; Sun et al., 2020). Particularly, acetyl coenzyme Arachidonic acid lipoxygenase (ALOX), cytochrome P450 oxidoreductase (CPY450), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and synthetase long-chain family 4 (ACSL4) contribute in the favorable control of ferroptosis. This is improved by certain modulations of the autophagic degradation pathway, causing peroxidation of lipids and promote iron deposits. In conclusion, ferroptosis is a type of lipid peroxidation that is dependent on iron and ultimately causes a disruption in the structure of the cell membrane, resulting apoptosis.
3.1 IronNearly all organisms contain iron, an important trace metal that contributes to various biological processes including metabolizing energy, nucleic acid production, and regeneration. Intracellular iron equilibrium, which is attained by an exacting control on absorption, transport, storage, and release mechanisms, is necessary to maintain the precise metabolism of iron. The small intestine's duodenum and upper jejunum are where iron ions are mainly absorbed. After escaping by intestinal luminal capillaries' endothelial cells, iron ions are attached to transferrin (TRF) in the plasma. They then attach to transferrin receptor 1 (TFR1) and are taken up by lattice protein endocytosis (Koleini et al., 2021). After iron uptake, it is reduced from trivalent to divalent iron in acidic endosomes via prostate hexagonal transmembrane epithelial antigen 3 (STEAP3) and discharged by divalent metal transporter protein 1 (DMT1) into the cell's cytoplasm for cellular usage (Luck and Mason, 2012). Unstable iron pools within cells can become saturated with excessive iron. Such pools of free divalent iron have the potential to induce ROS, particularly hydroxyl radicals, which are produced by the Fenton response. Polyunsaturated membrane lipids are damaged by deposited ROS, which impedes cellular activity and leads to cell destruction. When available iron exceeds biological requirements, cytoplasmic ferritin converts divalent iron into trivalent iron and stores it in ferritin complexes. Thus, ferritin abundance controls ferroptosis sensitivity: ferritin raises the susceptibility to ferroptosis when unstable iron pools are limited because it promotes iron ion preservation. In contrast, when ferritin is reduced, iron escapes into the unstable iron pool and improves ferroptosis susceptibility. Based on studies, the main mechanism by which iron is liberated from saturated ferritin is nuclear receptor coactivator 4 (NCOA4)-mediated directed autophagy. Iron moves into the cell's cytoplasm when ferritin is broken down in the lysosome, where it is transported by NCOA4 (Mancias et al., 2014; Yambire et al., 2019). By facilitating the release of excess iron into the outside of cells, the iron transporter protein 1 (FPN1) helps to improve in vivo homeostasis. High levels of FPN1 reduce iron-mediated ROS (Ward and Kaplan, 2012; Mancias et al., 2014). Ceruloplasmin (CP) also inhibits ferroptosis induced by erastin and GSH peroxidase 4 inhibitors (RSL3) by mediating iron efflux through binding to cell surface FPN1.
Iron-related cell death occurs when iron is overloaded. It's plausible that the oxidative possibility of iron, involving Fenton chemistry, initiates the production of peroxided lipids immediately (Shah et al., 2018). Increased levels of free iron can form unstable iron pools, and an increase in unstable iron pools in cells can cause membrane lipid peroxidation. There are two sources of unstable intracellular iron pools: one is ferritin's specific autophagy by NCOA4 liberates the iron ions from ferritin; the other is heme oxygenase-1 (HO-1), it promotes the breakdown of heme into ferric ions and is controlled with the nuclear factor erythrocyte 2-related factor 2 (NRF2) gene, leading to ex vivo and in vivo mitochondrial iron overload (Chang et al., 2018). Within the cell, excessive iron produces a high level of free radicals from oxygen, which causes peroxidation of lipids byproducts such as 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), and phospholipid hydroperoxides (PLOOH). These accumulating byproducts have the potential to induce ferroptosis and ultimately damage cells (Liang et al., 2023). Multiple other lab research demonstrate that ferroptosis is triggered when Fe2+ is used as a cofactor for ALOX or CPY450 enzymes, and that unstabilized iron, which does not bind to the enzymes, propagates these peroxides to cause the peroxidation of lipids (Wenzel et al., 2017; Shah et al., 2018). In addition, iron acts as the active center of lipoxygenase, which oxidizes polyunsaturated fatty acids on cell membranes through an iron-ion-dependent chain reaction, thereby promoting the proliferation of phospholipid peroxides and thus causing ferroptosis. Iron in the active center of lipoxygenase exhibits in the formation of toxic lipid peroxides. In brief, iron performs a dual function in ROS production and facilitates the synthesis of ALOX, which elevates lipid peroxide levels during ferroptosis. Under normal physiological conditions, iron autophagy maintains intracellular iron homeostasis. Over-activation of iron autophagy results in excessive intracellular iron deposition, causing glutathione peroxidase 4 (GPX4) levels to fall and GSH to become diminished, which finally causes cell membrane structures to collapse and rupture and iron death. In conclusion, iron excess causes the body's iron regulation to be upset, which results in ferroptosis.
Since iron is important to ferroptosis, the specific iron mechanism has attracted a new wave of attention. Iron-catalyzed lipid peroxidation to generate hydrogen peroxide products, iron-catalyzed cleavage reactions, and truncation of electrophilic reagent oxidation are the three main stages of iron-catalyzed reactions. In addition, other factors involved in iron regulation influence sensitivity to ferroptosis (Patel et al., 2019). Thus, GSH deficiency may promote peroxidation of lipids and provide free Fe2+ for the Fenton reaction, which may damage cell membranes and cause ferroptosis. Based on a new study, prominent protein 2 (PROM2) helps produce multivesicular bodies and exosomes that contain proteins that contain iron, which regulates the level of iron in the body. This mechanism promotes resistance to ferroptosis by facilitating the movement of iron ions outside the cell. Similarly, iron export mediated through FPN1 enhances resistance to iron death. Moreover, glutathione-iron complexes are temporarily formed during the process of absorbing iron. Ferroptosis is inhibited by the iron chaperone poly(rC)-binding protein 1 (PCBP1), which interacts with ferritin and injects Fe2+ into it (Stockwell, 2022).
3.2 Lipid peroxidation and ROSThe process by which oxidizing agents, such as non-radical compounds or liberated radicals, target lipids with double bonds between carbons is termed lipid peroxidation. In addition to causing direct harm to phospholipids, this attack acts as a signaling mechanism to initiate a process of cell death. PLOOH is highly biologically active and can cause damage to lipid bilayers of cell membranes (Catalá and Díaz, 2016). Numerous investigations showed that nonenzymatic phospholipid autoxidation or enzymatic phospholipid peroxides ALOX34,38,-40, and CPY450 cause lipid peroxidation (Yang et al., 2016; Wenzel et al., 2017; Chu et al., 2019; Li et al., 2021b). The membrane disruption results from the AA/AdA-PE-OOH formation using lipoxygenase-mediated peroxidation of lipids. Ferroptosis caused by RSL3 and erastin involves enzymes important for the lysophospholipid acylation pathway, including acyl-coenzyme A synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (Soupene and Kuypers, 2008; Shindou and Shimizu, 2009; Dixon et al., 2015). It found that ferroptosis may be suppressed in vitro and in vivo by pharmacologically or biologically disrupting the ACSL4-LPCAT3-ALOX cascade. Furthermore, CPY450-mediated lipid peroxidation serves as a substitute for ALOX in ferroptosis, facilitating polyunsaturated fatty acid (PUFA) peroxidation and causing cancerous cell death. Ferroptosis is not usually caused by prostaglandin peroxidase synthase 2 (PTGS2); instead, it is believed to as a biomarker. However, in neuronal cells following brain damage, PTGS2 might mediate ferroptosis. Non-enzymatic pathway products include malondialdehyde (MDA), isoprostane, and 4-HNE. MDA can bind to proteins and DNA to form cross-linked compounds, which can alter membrane structure and function (Zarkovic et al., 2013). Moreover, both AD and PD diseases are pathophysiological associated with increased intracellular MDA (Garcia et al., 2013). The regulation of many transcription variables involving Nuclear factor erythroid 2–related factor 2 (Nrf2) and peroxisome proliferator-activated receptor (PPAR), may stimulate 4-HNE, it can be promoted by various signaling mechanisms, such as those involving caspases and cell cycle controls (Dalleau et al., 2013). Neurodegenerative diseases are exacerbated by imbalanced redox equilibrium, which is brought on by elevated 4-HNE levels (Dalleau et al., 2013). However, the significance of Fenton chemistry and iron-dependent enzymes in initiating the peroxidation of lipids, results in ROS and ultimately causes ferroptosis.
A cascade reaction that occurs inside the organism, oxidative stress (OS) is primarily caused by a disruption in the redox equilibrium and an overabundance of ROS. Among such ROS are peroxides (O2·-), hydroxyl radicals (OH·-), peroxyl radicals (RO2·-), alkoxyl radicals (RO·-), and certain oxidizing agents/non-radicals, such as hydrogen peroxide (H2O2), ozone (O3), hypochlorous acid (HOCl), and singlet oxygen (1O2). The body could be harmed as a result of this exposure. However, the factors that contribute to the production of internal ROS include xanthine oxidase (XO), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), and the mitochondrial electron transport chain. Of these, the NOX family which consists of up of five monooxygenases (NOX 1-5) and two dioxygenases (Duox 1-2) produces the majority of ROS in the body (Wingler et al., 2011; Zhang R. N. et al., 2018). Nitrogen oxidase (NOX) produces ROS, including hydroperoxides (Bedard and Krause, 2007). Several studies have shown that a minimum of 3 NOX family members such as NOX1, NOX2, and NOX4 were identified to promote iron-dead cancer cell death (Xie et al., 2017; Yang et al., 2019, 2020). Dipeptidyl peptidase 4 (DPP4) attaches to NOX1 in colon tumors to initiate ROS formation, which encourages ferroptosis (Xie et al., 2017). Additionally, TP53 deletion inhibits the central production of DPP4, which permits membrane-associated DPP4-dependent peroxidation of lipids, ultimately causing ferroptosis (Xie et al., 2017). Mitochondria are the primary intrinsic source of reactive oxygen species (ROS). The electrons that are lost during oxidative phosphorylation in the electron transport chain of the mitochondrial membrane combine with molecules of oxygen to form the anion superoxide (O2·-). The voltage-dependent anion channel (VDAC) channel protein on the external mitochondrial membrane is in charge of transferring ions, ATP, and other metabolites into and out of the mitochondria. The main functions of VDAC are to regulate cell metabolism, maintain calcium intracellular homeostasis, and modify apoptotic processes (Skonieczna et al., 2017). Yagoda et al. finds that erastin reacts to VDAC2 effectively, allowing tumor cells to undergo ferroptosis. In the presence of oncogenic RAS, RAS-selective lethal small molecule 5(RSL5) interacts with VDAC 2/3 to generate ROS and activate death mechanisms (Yang and Stockwell, 2008). ROS controls apoptosis, proliferation, and other signaling pathways within cells. However, because of their high reactivity and thus elevated ROS, they react with nearly all kinds of biomolecules (Yagoda et al., 2007). Consequently, many cellular and extracellular components are susceptible to damage by increased and prolonged ROS amounts. For example, proteins and lipids may completely degrade or have permanent functional changes. The oxidization of lipids and proteins can result in genetic modifications and inflammatory reactions, and the development of diseases such as premature aging, functional loss, and neural death. Moreover, protein oxidation may promote the formation of insoluble protein aggregation. Many diseases have their molecular roots in such forms of protein aggregation such as Alzheimer's disease (Qin et al., 2006) and PD (Zhang et al., 2000).
3.3 PUFAsFatty acids are processed for multiple purposes, including the synthesis of energy, cell membrane development, and a variety of signaling molecules. They perform a significant part in the cellular metabolism of lipids (Patel et al., 2019). The fatty acids can be divided into three main types: monounsaturated fatty acids (MUFAs, 1 double bond), saturated lipids (no double bonds), and polyunsaturated Lipids (PUFAs, >1 double bond). Under physiological conditions, PUFAs enhance human metabolism and promote fat removal. As components of cell membranes, many biological processes, including as inflammatory processes, immunology, synaptic plasticity, and cell development, are mediated by PUFAs (Guzik et al., 2000). Lipidomics studies have shown that the phospholipids (PLs) of the most vulnerable lipids to peroxidation are PUFAs and cell death. The diallyl carbons (carbon atoms adjacent to two neighboring carbon-carbon double bonds) in PUFA-PLs are chemically susceptible to attack by free radicals, lipoxygenases (LOXs), and ROS, and thus serve as key sites in the lipids that drive ferroptosis (Zorov et al., 2014). Phospholipid-containing PUFAs can form lipid H2O2 by enzymatic (most notably LOXs) or non-enzymatic (Fenton chemical) oxidation reactions, which bind to iron to produce toxic lipid free radicals that lead to cellular damage. In vivo, the most common PUFAs is AA/AdA, which is present in all tissues. The formation of these PUFAs coenzyme A derivatives and their insertion into phospholipids is required for the generation of ferroptosis signals. AA/AdA-PE biosynthesis requires the involvement of ACSL4 and LPCAT3. Thus, deletion of the AA/AdA-PE product depletes the substrate for lipid peroxidation and increases resistance to ferroptosis (Garcia et al., 2013; Dixon et al., 2015; Doll et al., 2017). Cells supplemented with AA or other PUFAs are sensitive to ferroptosis (Yang et al., 2016). Studies have shown that the peroxide site of PUFAs contains deuterium, which can slow the oxidation of PUFAs and inhibit ferroptosis. Deuteration at AA-7 has a protective effect against ferroptosis induced by RSL3 (Gaschler et al., 2018). Thus, the state of PUFAs influences lipid peroxidation. In addition, GPX4 inhibitors can cause hyper oxidation of PUFAs, so the effectiveness and synthesis of PUFAs is a good target for modulating sensitivity to ferroptosis, and modulation of PUFAs may be a good approach for treating diseases associated with ferroptosis. Figure 1 shows the mechanism of ferroptosis. Tables 1, 2 summarize the inducers and inhibitors of ferroptosis.
Figure 1. The ferroptosis mechanism involves iron, ROS, and lipid peroxidation.
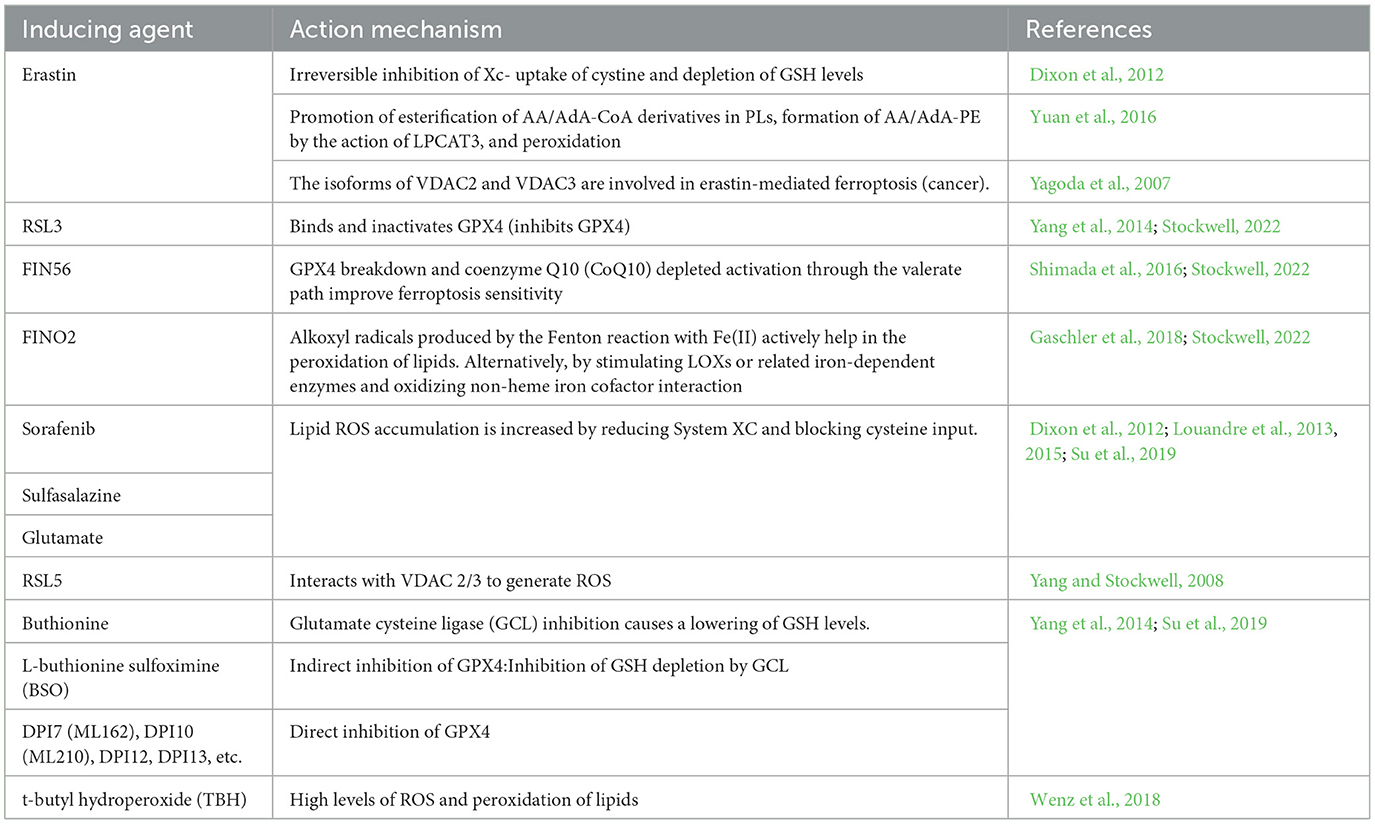
Table 1. Inducers of ferroptosis.
Table 2. Inhibitors of ferroptosis.
4 Organelles related to ferroptosis 4.1 LysosomesIt has been shown that the lysosome is an important intracellular storage site for iron ions, which means that it is essential to iron regulation and metabolic activity (Yuan and Ofengeim, 2023). When intracellular levels of iron ions are too high, iron can accumulate in lysosomes, helping in protecting tissues against the negative effects of iron overload (Wang J. et al., 2023). However, ferroptosis may also be triggered when there is too much iron in the lysosome. Second, lysosomes are involved in the release and reuse of iron through their internal acidic environment and abundance of hydrolytic enzymes (Hung et al., 2023). Under specific conditions, such as oxidative stress or cellular injury, the permeability of the lysosomal membrane may increase, causing iron to be released into the cytoplasm from the lysosome. The release of iron may worsen cellular damage and ferroptosis by promoting peroxidation of lipids and the ROS. In addition, as mentioned earlier, lysosomes and autophagy are closely related processes. The mode of intracellular self-digestion identified as autophagy breaks down and recycles intracellular components by encasing intracellular material in autophagic vesicles and fusing with lysosomes (Li et al., 2023). During ferroptosis, autophagy may be activated, resulting in more iron being transported into the lysosome. This further exacerbates iron overload in lysosomes and promotes ferroptosis (An et al., 2023).
Even so, multiple research suggests that by promoting the activity of the antioxidant gene superoxide dismutase 1 (SOD1), activating the nuclear transcription factor EB (TFEB) may prevent lysosome-based ferroptosis. It suggests that there is interaction between the nucleus and the lysosome, which may establish a feedback loop to control ferroptosis (Cai et al., 2024; Calabrese et al., 2024; Shariq et al., 2024). Nevertheless, more research are needed on whether lysosomal cytosolic action leads to membrane remodeling and repair during ferroptosis.
Overall, lysosomes play an integral part in ferroptosis by storing, releasing, reusing, and interacting with processes such as autophagy. Together, these complex processes and mechanisms determine the fate of cells under conditions including iron overload or oxidative damamge. The particular functions and processes that control lysosomes in ferroptosis remain mainly unresolved, necessitating more research and study.
4.2 MitochondriaThe main ROS contributor in cells, especially after oxidative phosphorylation is the mitochondria. Localized ROS production not only causes immediate damage to the mitochondria but also modifies the cell's overall oxidative state (Jia et al., 2024a). Research indicates that increased ROS in the mitochondria generates ferroptosis, which can be impeded by antioxidants or enzymes that target the mitochondria (Chen et al., 2024). Secondly, ferroptosis is inhibited partly by certain antioxidant enzymes identified in mitochondria. For example, GPX4 can be identified in the gaps between the cytoplasmic and mitochondrial membranes, and its structure reduces the oxidative stress that destroys mitochondria after a cell dies (Guo et al., 2024).
By living within the matrix of mitochondria, members of the ferromanganese SOD2 may be able to inhibit ferroptosis in non-small cell lung cancer (NSCLC) cells prompted by mitochondrial ROS (Zhao et al., 2023). Moreover, by interacting with ALOX5, microsomal glutathione S-transferase 1 (MGST1) suppresses the ferroptosis and peroxidation of lipids (Zhang et al., 2023).
Moreover, mitochondria control iron metabolism, which affects ferroptosis. The balance of cellular metabolism of iron depends on mitochondria, becoming a significant organelle. By increasing the quantity of iron stored in the mitochondria and lowering cellular ROS levels, overexpression of mitochondrial ferritin can prevent ferroptosis (Xu et al., 2024).
Moreover, the process of apoptosis produced by cysteine deficiency involves mitochondria. The lack of cysteine causes the potential of mitochondrial membranes to become highly polarized and lipid peroxides formation, which finally results in ferroptosis (Du et al., 2024). However, increased polarization of the mitochondrial membrane potential, peroxide-lipid formation, and ferroptosis are lowered when the electron transport chain (ETC) or the mitochondrial tricarboxylic acid cycle (TIC) is inhibited. In conclusion, mitochondria have an important part in ferroptosis through ROS production, the activity of antioxidant enzymes, the control of iron metabolism, and cysteine deprivation. As a result, mitochondria have significance to the ferroptosis process.
4.3 ERThe main site for protein production and use in the cell, the Endoplasmic Reticulum (ER), has significance for maintaining cell equilibrium because it contains a significant quantity of membrane lipids. Despite the several stresses, ferroptosis is one to which the ER reacts significantly. Stress in the upper respiratory tract (ER) can set off an unfolded protein process that attempts to restore the proliferative equilibrium. If a cell cannot regain homeostasis, ER stress can lead to cell loss, including ferroptosis (Wang et al., 2024). Second, the peroxidation of lipids in ferroptosis is mainly regulated by the ER. As evidenced by the finding that Fer-1 occurs primarily in the ER, the ER is most important for the peroxidation of lipids during ferroptosis. ER viscosity increases during ferroptosis, possibly due to the aggregation of PUFA-PLs leading to the hardening of the ER (Stockwell, 2022). Ferroptosis is characterized by a substantial anticipation of lipid peroxidation. In addition, significant functions are played by specific ER proteins in controlling ferroptosis susceptibility.
For instance, a transmembrane protein, Stimulator Of Interferon Response CGAMP Interactor 1 (STING1) on the ER translates oxidative responses in the nucleus or mitochondria into a ferroptosis response. STING1 is ligand-activated in response to the oxidized nucleic acid base 8-hydroxyguanine released by iron-dead cells activated, thereby triggering ferroptosis (Hirschenberger et al., 2023). It indicates that STING1 regulates cellular ferroptosis in a variety of mechanisms. In addition, endoplasmic reticulum stress-associated calcium ion efflux triggers the endocytosis-associated complex Endosomal Sorting and Transport Complex-III (ESCRT-III) to accumulate in the plasma membrane to prevent membrane damage during ferroptosis (Lin et al., 2023). This suggests that during ferroptosis, the ER is crucial for preserving the functionality of the cell membrane and averting damage to the membrane. Overall, the ER role in ferroptosis comprises multiple aspects of protein synthesis and processing, lipid peroxidation, regulation of specific proteins, and maintenance of membrane integrity, suggesting that the ER has significance to ferroptosis.
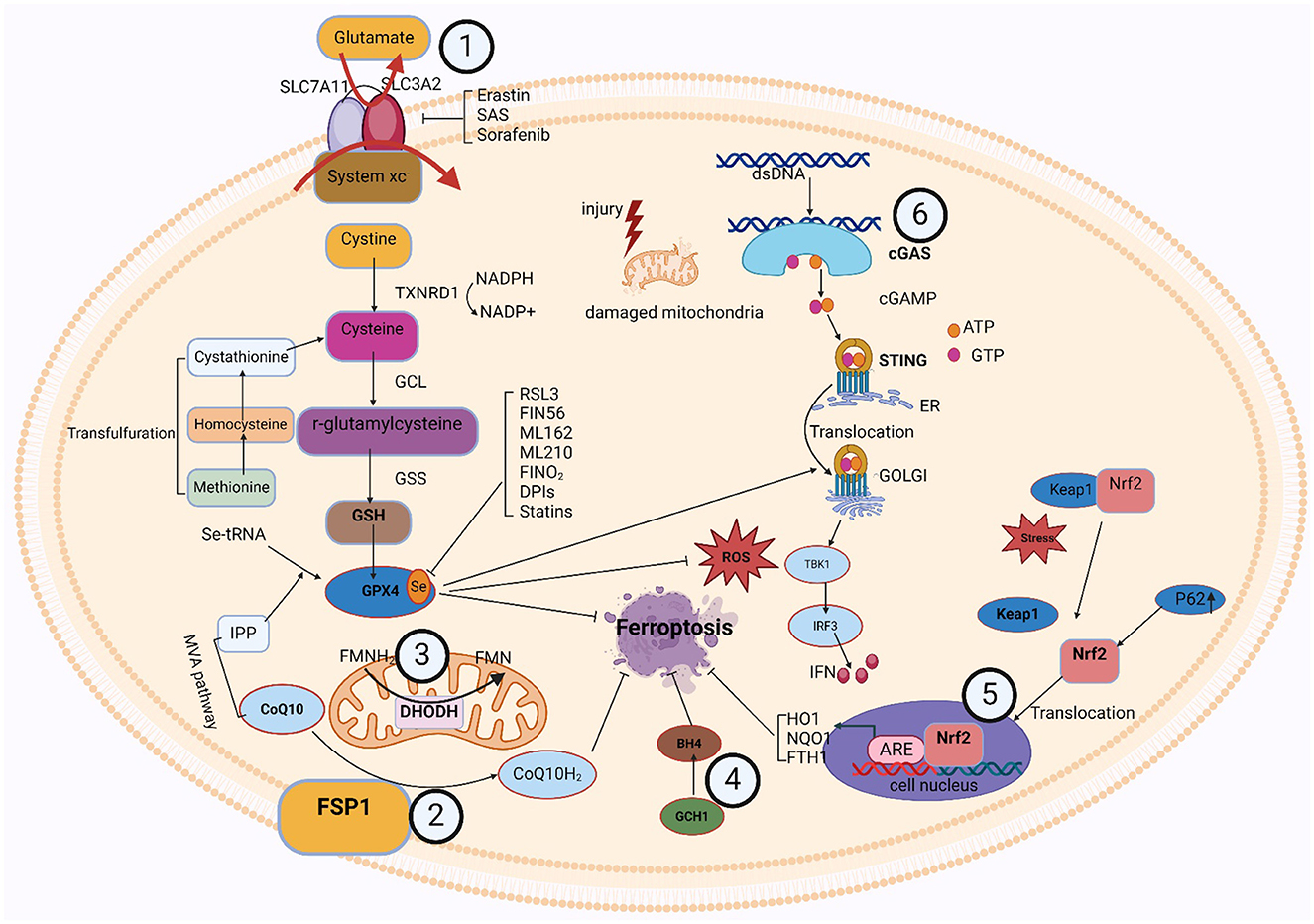
5 Metabolic pathway and signaling pathways related to ferroptosis 5.1 GPX4-GSH metabolic pathwayThe formation of glutathione (GSH) and the activity of glutathione peroxidase 4 (GPX4) are important components of this pathway. Cysteine and glutamate transfer throughout the cell membrane and GSH synthesis relies on the Xc-antioxidant system, comprised of the SLC7A11 and SLC3A2 bis-subunits (Chu et al., 2019). When cystine is taken up by the xc-system, GSH reductase reduces it to cysteine. The thioredoxin reductase 1 (TRXR1)/thioredoxin 1 (TRX1) system also maintains a reduced pool of GSH without GSH reductase (Prigge et al., 2017). Cysteine is a nonessential amino acid, and under certain conditions, methionine can be converted via the transsulfuration pathway to homocysteine, cystathionine, and ultimately cysteine (Dixon et al., 2015; Zou et al., 2020; Yan et al., 2021). Suchprocess is subject to the cysteine-tRNA synthetase I being negatively regulated (Hayano et al., 2016). Thus, inhibition of the transsulfuration pathway is expected to increase sensitivity to ferroptosis. The cytoplasmic enzymes glutathione synthetase (GSS) and GCL mediate the conversion of glycine, glutamate, and cysteine into GSH. Additionally, GPX4 contributes to detoxification by accelerating the transformation of H2O2 and hydroperoxides into water or ethanol (Bela et al., 2015), relying on the effectiveness of glutathione as a cofactor. In addition to its major cofactor GSH, GPX4 demonstrates versatility by employing various low-molecular thiols, including protein thiols. Selenoprotein GPX4's active site contains selenocysteine (Sec). Additionally, the synthesis of selenoproteins, such as GPX4, is regulated by the mevalonate (MVA) pathway. The biological activity of GPX4 and the synthesis of GSH collaborate to inhibit ferroptosis in cells at a molecular level (Soupene and Kuypers, 2008). Erastin, Due to their specific inhibition of the Xc-system, sorafenib and sulfasalazine (SAS) suppress GPX4 directly by depleting GSH, while RSL3 effectively blocks GPX4 function (Yang et al., 2014). Inhibiting intracellular cysteine and GSH levels affects GPX4 function since cysteine is the rate-limiting precursor for GSH formation and GSH is a major antioxidant in mammalian tissues, thereby making cells more susceptible to ferroptosis (Soupene and Kuypers, 2008).
It has been shown that there are interactions between various organelles and the GPX4-GSH metabolic pathway. For the intracellular interaction between lysosomes for cells to remain in a state of redox equilibrium and to avoid ferroptosis, the GPX4-GSH metabolism process is mandatory. The primary intracellular digesting and recycling organelles, lysosomes are in charge of breaking down and recycling a range of macromolecules, such as degraded fatty acids, and nucleic acids. In the GPX4-GSH metabolic pathway, lysosomes may regulate GPX4 activity by degrading damaged GPX4 or affecting its upstream molecules (Deng et al., 2023). In addition, iron ions released by lysosomes may indirectly affect GPX4 activity because to scavenge lipid peroxides, GPX4 requires GSH as a reductant, and iron ions may affect the synthesis or utilization of GSH. The GPX4-GSH metabolic pathway is also capable of affecting lysosomes. Ferroptosis can be avoided by scavenging lipid peroxides that are activated with iron ions, owing to the important antioxidant enzyme known as GPX4 (Xue et al., 2023). GPX4 requires GSH as a reductant to fulfill its function. In the GPX4-GSH metabolic pathway, GPX4 activity may directly affect lysosomal stability and function. For example, when GPX4 activity is reduced, the intracellular lipid peroxidation level is elevated, which may result in damage to the lysosomal membrane, thereby affecting the normal function of lysosomes. The maintenance of lipid peroxidation levels, protein synthesis, and cellular redox equilibrium depends on the interactions between the ER and the GPX4-GSH metabolic pathway. In addition to being linked to the formation of lipids and metabolism, the ER is the primary site for protein production and modification in cells. GPX4 can scavenge lipid peroxides catalyzed by iron ions, thereby preventing ferroptosis. GPX4 requires GSH as a reductant to perform its function (Jia et al., 2024b). The ER supports the activity of GPX4 by synthesizing and supplying antioxidants, such as GSH, thereby helping cells to resist peroxidation of lipid and ferroptosis. Moreover, the production and function of GPX4 are controlled by the ER and modulates its antioxidant capacity by affecting the post-translational modification or stability of GPX4. The GPX4-GSH metabolic pathway positively affects ER function by scavenging lipid peroxides and maintaining cellular redox homeostasis (Chen et al., 2017). Lipid peroxidation damages the ER membrane and the proteins therein, leading to ER stress and dysfunction. By scavenging lipid peroxides, GPX4 can reduce the damage to the ER and maintain its normal function. In cell metabolism and antioxidant defense systems, there is a significant interaction between the GPX4-GSH metabolic process and mitochondria. The important internal organelles known as mitochondria are in control of generating energy and engaging in a variety of metabolic activities. Within GPX4-GSH metabolic pathway, mitochondria provide reduced GSH to GPX4 as a reductant essential to its antioxidant activity. GSH, through the action of GPX4, can scavenge phospholipid hydroperoxides catalyzed by ferric ions, thereby preventing ferroptosis. Therefore, the mitochondrial function has a direct impact on the antioxidant capacity of the GPX4-GSH metabolic pathway, which also affects the mitochondria. by scavenging phospholipid hydroperoxides catalyzed by ferric ions, the GPX4-GSH metabolic pathway prevents lipid peroxidation damage to the mitochondrial membrane, thus maintaining the mitochondrial functional and structural integrity (Asperti et al., 2021). Furthermore, the GPX4-GSH metabolic pathway can lessen OS-induced mitochondrial destruction by scavenging ROS in mitochondria (Yuan et al., 2021). Consequently, the GPX4-GSH pathway's activation is required for the mitochondria to function normally. In summary, there is an interaction between various intracellular organelles and the GPX4-GSH metabolic pathway. This interaction is important for maintaining cellular redox homeostasis and preventing lipid peroxidation and ferroptosis. Thus, thorough research on the relationship between organelles and the GPX4-GSH metabolic pathway could yield important insights into the management or avoidance of ferroptosis-related disorders.
5.2 FSP1-CoQ/DHODH-CoQ/GCH1-BH4/Nrf2-Keap1 signaling pathwayIn the last few years, three GPX4-independent signaling pathways have been identified to inhibit ferroptosis: Ferroptosis Suppressor Protein 1 (FSP1)/CoQ10, Dihydroorotic acid dehydrogenase (DHODH), and GTP Cyclic Hydrolyserase 1 (GCH1)/Tetrahydrobiopterin (BH4). Bersuker et al. (2019) and Doll et al. (2019) discovered the CoQ10-FSP1 route, an innovative ferroptosis inhibition pathway. At cell membrane, FSP1 behaves as an oxidoreductase to promote NADPH oxidase-catalyzed CoQ10 to generate CoQ10H2 (Shimada et al., 2016). This pathway synergizes with GSH-GPX4 to inhibit ferroptosis.
Two more GPX4-independent systems that prevent lipid peroxidation and ferroptosis are reported in 2020 and 2021. In 2020, it was found that GCH1 regulates the levels of PLs through two PUFAs tails to produce the lipophilic antioxidant BH4, defends cells against ferroptosis (Kraft et al., 2020). The first defense mechanism against DHODH-mediated mitochondrial ferroptosis was initially shown by Boyi's group in 2021. To avoid ferroptosis in the inner membrane of the mitochondria, DHODH lowers CoQ to CoQH2. This reaction occurs in tandem with mitochondrial GPX4 (Mao et al., 2021). Ferroptosis was unable to affect cells with high levels of GCH1 or DHODH, and was more probable to affect cells with low expression. Antioxidant proteins are produced under the control of Nrf2, which protects cells from oxidative damage. Kelch-like ECH-associated protein 1 (Keap1) controls its activity. When Keap1 attaches to Nrf2 in the cytoplasm, Nrf2 becomes ubiquitinated and is then degraded by the proteasome (Song and Long, 2020). Nrf2 is translocated to the nucleus and released from the Keap1 binding site in response to oxidative damage. To stimulate the transcription of protective antioxidant genes, Nrf2 can interact with antioxidant response elements (AREs) in the nucleus (Zhang, 2006). Autophagy has been a regulatory mechanism for these activities. By deactivating Keap1, the multipurpose protein p62, which is found in the cell and acts as an autophagy receptor, stimulates Nrf2 (Komatsu et al., 2010). The p62-Keap1-Nrf2 antioxidant signaling pathway is responsible for the inability of hepatocellular carcinoma (HCC) cells to enter ferroptosis, as revealed by Tang's group in 2016 (Sun et al., 2016). The p62-Keap1-NRF2 pathway was shown to be significant in defending HCC from ferroptosis. Multiple genes involved in the breakdown of iron and ROS, such as quinone oxidoreductase-1 (NQO1), heme oxygenase-1 (HO1), and ferritin heavy chain 1 (FTH1), are stimulated to produce that protective effect (Sun et al., 2016). In addition, the genes SLC7A11 and GSS, which encode GSH synthesizing proteins, are also considered Nrf2 targeted genes (Sun et al., 2016). These findings imply that Nrf2 is crucial for ferroptosis resistance.
It has been shown that there are interactions between various organelles and the signaling path between Nrf2-Keap1. Lysosomes and the Nrf2-Keap1 signaling pathway interact in a complicated and important way, and is important for regulating cellular redox balance and shielding cells from xenobiotic invasion and oxidative damage. By attaching to and increasing the breakdown of Keap1, a significant Nrf2 inhibitor that keeps Nrf2 levels low, lysosomes modify the processes of the Nrf2-Keap1 pathway (Amorim et al., 2022). Lysosomes can degrade Keap1 through their internal acidic environment and abundance of hydrolytic enzymes, thereby relieving the inhibitory effect on Nrf2. When lysosomal function is impaired or subjected to certain stimuli, the degradation of Keap1 may be blocked, leading to the accumulation and Nrf2 stimulation. Lysosomes are the subject of the Nrf2-Keap1 activation pathway. By controlling the levels of antioxidant proteins and detoxify enzymes, the Nrf2-Keap1 signaling pathway preserves cellular redox equilibrium. They protect the lysosomal membrane from oxidative damage and maintain lysosomal stability and function. In addition, activation of Nrf2 promotes autophagy (Meeran et al., 2021). Therefore, the Nrf2-Keap1 signaling pathway can affect lysosomal function and activity by regulating autophagy. The interaction between ER and the Nrf2-Keap1 signaling pathway is important for maintaining cellular homeostasis, coping with oxidative stress, and regulating protein synthesis and degradation. ER is not only a major site of protein synthesis and modification but also takes part in several signal transduction pathways (Cong et al., 2021). In the Nrf2-Keap1 signaling pathway, ER may regulate the activity of Nrf2 by affecting the modification and stability of Keap1. For example, certain enzymes in the ER may modify Keap1 and affect its ability to bind to Nrf2, thereby regulating the stability and transcriptional activity of Nrf2. In addition, ER stress may also affect the activity of the Nrf2-Keap1 signaling pathway, which enhances cellular antioxidant capacity by inducing the degradation of Keap1 or promoting the nuclear translocation of Nrf2. On the other hand, the Nrf2-Keap1 signaling pathway maintains cellular redox homeostasis by regulating the expression of antioxidant proteins and detoxifying enzymes. These antioxidant proteins and detoxifying enzymes protect the ER from damage by oxidative stress and maintain its normal function (Park et al., 2016) The Nrf2-Keap1 signaling system and mitochondria interact to a great extent in cell metabolism, redox homeostasis, and antioxidant defense. One of the primary contributors of intracellular ROS is superoxide, which is formed in the mitochondrial oxidative respiratory chain. The Nrf2-Keap1 pathway's activity may be affected by certain ROS. The thiol moiety of Keap1 may be oxidized in the presence of high ROS levels, resulting in a conformational shift that releases Nrf2 (Masuda et al., 2024). To preserve cell redox equilibrium, the emitted Nrf2 subsequently reaches the nucleus and starts the transcription of antioxidant genes. Furthermore, modulation of the Nrf2-Keap1 process may raise the synthesis of certain enzymes that detoxify and antioxidant proteins that can scavenge ROS and prevent oxidative stress-induced mitochondrial injury. In addition, some antioxidant proteins can be directly localized in mitochondria, such as mitochondrial thioredoxin and mitochondrial GPX, which are directly scavenging ROS, preserve mitochondrial structure and function (Sun et al., 2024). In summary, there is an interaction between various intracellular organelles and the Nrf2-Keap1 pathway. This interaction is important in preventing lipid peroxidation and ferroptosis, among others.
5.3 cGAS-STING signaling pathwayThe cGAS-STING [(cGAMP) synthase (cGAS)-interferon gene-stimulating factor (STING)] signaling pathway is a mediator of the inflammatory response under stress, infection, and tissue injury. Cell-microbe and host-derived double-stranded DNA (dsDNA) can be sensed and regulated for defense against extracellular or intracellular infection (Hopfner and Hornung, 2020; Decout et al., 2021). Then, the production of the second messenger (cGAMP) is catalyzed by cGAS (Cheng et al., 2020). Subsequently, STING bound to cGAMP oligomerizes at the endoplasmic reticulum membrane and then translocates from the ER to the Golgi compartment, mediating tank-binding kinase 1 (TBK1) autophosphorylation (Cheng et al., 2020). As a result, TBK1 phosphorylates STING, which then attracts interferon (IFN) regulatory factor 3 (IRF3) to continue phosphorylation (Cheng et al., 2020). IFN-stimulated genes (ISGs) and type I IFN transcription are mediated by phosphorylated IRF3 dimers, which are moved to the nuclei and engaged in an array of pathological events (Jiang et al., 2014a,b; Negishi et al., 2018; Cheng et al., 2020).
Studies have revealed an association between the cGAS-STING process and ferroptosis. Various studies attested to STING's role in the ferroptosis mechanism. According to Li et al., mitochondrial oxidative damage was brought on by erastin, an established inducer of ferroptosis. Moreover, mitochondrial STING expedited the mitochondrial fusion that depends on the mitofusin gene (MFN1/2), which leads to ferroptosis and peroxidation of lipids (Li et al., 2021a). Meanwhile, by decreasing the vulnerability of cells to ferroptosis through the inhibition of STING or MFN1/2, erasin's tumor-suppressive effects were reduced (Li et al., 2021a). Furthermore, IRF3 is involved in ferroptosis and peroxidation of lipids as an alternative target of STING. Docosahexaenoic acid (DHA) decreased ALOX activity, raised IRF3 expression, aided SLC7A11 transcription, and halted ferroptosis in a mouse model of cardiac hypertrophy carried on by angiotensin II (Shi et al., 2022). There could be an effect of ferroptosis on the cGAS-STING regulation pathway. Jia et al. (2020) found that the stimulation of the cGAS-STING signaling mechanism was hampered by an increased cell peroxidation of lipids brought on by GPX4 knockdown. Moreover, ferroptosis in mice brought on by a high-iron diet or a GPX4 loss is shown to stimulate kras-mediated pancreatic tumor formation, based on research by Dai et al. (2020). Integrating results suggests that ferroptosis and lipid peroxidation are related to the active cGAS-STING regulation mechanism, and that iron prolapse's detrimental effects can be lessened by concentrating on this mechanism.
It was discovered that numerous organelles engage with the cGAS-STING signaling cascade. The association between lysosomes and the cGAS-STING signaling cascade has gradually gained more attention in recent years. The implications of this association extend to cellular immunity and signaling. In addition to breaking down a variety of macromolecules, lysosomes are intracellular recycling and digesting organs that may also affect the activity of the cGAS-STING cascade (Gaidt et al., 2017). First, lysosomes may negatively regulate the activity of the pathway by degrading cGAS or STING proteins. Second, iron ions or other molecules released by lysosomes may directly affect the enzymatic activity of cGAS or the phosphorylation state of STING, thereby modulating the activity of the pathway. However, lysosomes are also affected by the cGAS-STING signaling mechanism. When the cGAS-STING pathway detects intrinsic DNA damage or DNA viruses, it initiates a cascade of signaling events that trigger the synthesis of inflammatory mediators and the recruitment of lymphocytes. This activation state may indirectly affect lysosomal function. For example, activated immune cells may enhance bactericidal or anti-tumor capabilities by releasing lysosomal enzymes (Wu et al., 2024). Furthermore, by controlling the expression of specific transcription variables, the cGAS-STING signaling network may have an impact on lysosomal formation and function the interaction between ER and the cGAS-STING signaling pathway plays an important role in cellular immunity and antiviral responses. ER is not only a major site of protein synthesis and modification but also participates in a variety of signal transduction processes. The ER offers an area for the cGAS to react in the cGAS-STING signaling process, allowing the cGAS to identify and engage DNA in the cell's cytoplasm before activating and synthesizing the second messenger, cGAMP. Through its unique membrane structure, cGAS-STING also gives STING an anchor site, allowing STING to locate and operate on the ER membrane (Thomsen et al., 2024). cGAS-STING signaling pathway is also capable of acting on the ER. The second messenger, cGAMP, is produced by cGAS upon recognition and binding to DNA in the cytoplasm, activating STING. From the ER, active STING travels to the Golgi apparatus, where it recruits and activates signaling molecules downstream, including IRF3 and TBK1. Type I interferon is produced and secreted as a result of these communication pathways, and this initiates an antiviral immune response (Gong et al., 2024). The structure and function of ER may be affected in some way during this process, such as induction of ER stress and alteration of protein synthesis. The key component of cellular immunity and the antiviral response is played by the association between mitochondria and the cGAS-STING regulating cascade. As significant intracellular organelles, mitochondria not solely serve as the main site for generating energy but also take part in several different cell signaling endeavors. Mitochondria may activate cGAS in the cGAS-STING signaling circuit by producing ROS. The amount of ROS rises in mitochondrial infection or damage. These ROS can trigger cGAS, which may then attach and identify DNA in the cytoplasm, activating and synthesizing cGAMP, and cGAS will attach to DNA in the cell cytoplasm after recognizing it. Upon identifying and attaching itself to cytoplasmic DNA, cGAS produces another messenger, cGAMP, which then triggers STING to initiate a sequence of transmitted signals cascades (Jiménez-Loygorri et al., 2024). This series of signaling processes leads to the synthesis and secretion of type I interferon, which in turn triggers an antiviral immune response. During this process, mitochondria may be affected in certain ways, such as changes in mitochondrial function and fluctuations in ROS levels. Moreover, the mitochondrial autophagy process may be impacted by the initiation of the cGAS-STING method, which maintains cellular homeostasis by removing damaged or excess mitochondria (Zhou et al., 2024). In summary, there is an interaction between various intracellular organelles and the Nrf2-Keap1 pathway. This interaction is important in preventing lipid peroxidation and ferroptosis, among others. Figure 2 shows the signaling pathways associated with ferroptosis.
Figure 2. The signaling pathways associated with ferroptosis.
6 Pathologic relationship between ferroptosis and neurodegenerative diseasesThe hallmark of neurodegenerative diseases is the gradual death of neurons and are brought on by an accumulation of oligomers and aggregates of misfolded proteins (Menzies et al., 2017; Djajadikerta et al., 2020). Ferroptosis has been linked to several mechanisms that occur in neurological disorders, including peroxidation of lipids, GSH depletion, and brain depositing iron (Reichert et al., 2020). Moreover, ferroptosis in motor dementia was linked to reduced functioning of the GSH and GPX4 networks (Johnson et al., 2012; Dias et al., 2013; Blesa et al., 2015; Gu et al., 2015). For healthy neural development and mental ability, iron is important. With age, higher amounts of iron are observed in the caudate nucleus, pallidum, shell nucleus, substantia nigra, and other basal ganglia and cortex regions. Certain areas of the body accumulate iron, which is associated with neurological disorders. Iron overload in the brain can lead to GPX4 deficiency and lipid peroxidation in neurons and glial cells. Research has identified an association between motor nerve deterioration and increased iron (Chen et al., 2015; Reichert et al., 2020). Deposits of iron in specific brain regions can hasten the death of neurons and glial cells in neurological disorders. The inflammation of the brain is marked by immune cell infiltration into areas with small lesions, and this process is frequently linked with brain inflammation (Urrutia et al., 2021). Under a microscope, the primary sites of brain iron formation are dystrophic microglia invaded by astrocytes and iron-containing macrophages. Recent work shows that increased cytoplasmic levels of iron can cause microglia to polarize toward an inflammatory M1 phenotype (Kroner et al., 2014). It was proposed that the neuroinflammatory reaction can be affected by the polarization of microglia (Cherry et al., 2014). Iron increases into deep gray matter and cortical astrocytes and microglia as individuals age, but it stays constant in oligodendrocytes (Connor et al., 1990). Usually, iron accumulates in oligodendrocytes, where it is primarily stored as ferritin and transferrin. These levels of iron remain unchanged with aging. Oligodendrocytes are the primary source of iron and irons-binding proteins including ferritin and TRF. Cell damage can be triggered immediately by iron deposits in neurons (Williams et al., 2012). The potential medicines to inhibit the development of AD and PD have been proposed, including ferritin 1 and iron chelators such as Deferoxamine (DFO), M30, and lipoic acid. These substances may work as protectors by making hypoxia-inducible factor 1α (HIF1α) in the brain more stable. The tissue of the brain is rich in PUFA lipids, including arachidonic acid (AA) and docosahexaenoic acid (DHA), which also has an increased lipid content, fewer antioxidant enzymes, and higher oxidative metabolism (Olmez and Ozyurt, 2012; Larrieu and Layé, 2018). In the brain's tissue, peroxidation of lipids becomes accessible. Multiple studies have found that increased intracellular liberated radical oxidative damage linked to iron metabolism is part of the cause of neurological diseases (Stockwell et al., 2017; Mao et al., 2020). Because ferroptosis exerts a role in the pathomechanism of neurodegenerative diseases, targeting it could provide therapeutic opportunities for their treatment.
6.1 Ferroptosis and ADAlzheimer's disease (AD) is a common neurodegenerative condition in older adults that focuses primarily on the hippocampal and cerebral cortex. The AD pathogenesis is typified by the development of Tau protein-containing neurofibrillary knots and senile plaques, which are composed of Aβ polypeptides (Serrano-Pozo et al., 2011; 2022). Genetic alterations in AD include changes in the genes encoding amyloid precursor protein (APP), apolipoprotein E (ApoE), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) (Scheltens et al., 2021). APP cleaves to form amyloid beta (Aβ), while hyperphosphorylation leads to the separation and aggregation of Tau proteins from microtubules, creating tangles in nerve fibers of nerve cells. Physiologically, APP enters the non-amyloid pathway via alpha-secretase and gamma-secretase cleavage. Amyloidosis arises from the aberrant cleavage of APP by β- and γ-secretases, releasing neurotoxic fragments such as Aβ40 and Aβ42, which are capable of aggregating and subsequently developing plaques that contribute to responsible for AD (Yan and Zhang, 2019). Mutations in PSEN1 and PSEN2, part of the catalytic protease compound that precisely cleaves APP and other proteins, and are linked to AD with early manifestations (Walter, 2015). The plasma membrane of neurons is rich in PUFAs and is susceptible to free radical attack and peroxidation of unsaturated carbon-carbon bonds. Pathogenesis of the nervous system is related to ROS (Yang and Stockwell, 2008).
Recent evidence suggests that Ferroptosis may play an integral part in AD development. In the brain, iron is a biological element that is vital to several significant processes. The synthesis of neurotransmitters, myelin formation, oxidative phosphorylation, and transport of oxygen are some of these processes (Ward et al., 2014). Recent research exists to support the direct induction of ferroptosis by perturbations in iron regulation, both in vivo and in vitro (Wang et al., 2017). Iron-storing proteins and Cp were shown at higher levels in AD patients than in healthy people, based on Ashraf et al. However, it has been shown that AD sufferers express DMT1 and FPN1 at reduced levels (Ashraf et al., 2020). Age-related iron formation was shown by Raven et al. (2013) to be a factor in damage to tissues, cognitive loss, and clinical AD manifestations. Neuronal toxic levels decreased substantially when Aβ had been treated by using the iron chelator DFO (Rottkamp et al., 2001). In rats, amyloid-beta deposition and tau excess phosphorylation have been exacerbated by a high-iron meal; whereas, iron chelators successfully decreased amyloid beta deposition and tau excess phosphorylation (Sripetchwandee et al., 2014). In the APP/PS1 mutant mice species, Svobodová et al. (2019) the formation of amyloid plaques in the brain cortex corresponds to the accumulation of ferritin and liberated iron. Additionally, It is already known that iron accumulates in the body and leads to the abnormal formation of Aβ and neurofibrillary tangles (NFTs), resulting in Aβ plaque misfolding (Yan and Zhang, 2019). Extensive experimental studies showed that the beta-amyloid interacts with iron, leading to Aβ to clump together and become cytotoxic (Bousejra-ElGarah et al., 2011; Lermyte et al., 2019). In contrast, iron has an eight-fold higher binding affinity for Aβ than TRF, thereby interfering with iron homeostasis (Jiang et al., 2009). The aggregation of proteins associated with tau is a significant hallmark of AD. Iron induces and regulates tau protein phosphorylation (Nikseresht et al., 2019; Yan and Zhang, 2019). Wan et al. (2019) found that as compared to control group, neurons exposed to ferrous iron displayed 110% higher phosphorylated tau protein at S396. Amyloid precursor protein (APP) was recently identified by Tsatsanis et al. (2020) to regulate the cellular-surface expression of membrane-bound iron transporters, hence improving neural iron efflux. Tau deficiency renders APP-mediated iron export impure, causing an increase of iron inside cells. Furthermore, the advancement of AD and cognitive impairment has been associated with increased brain iron concentrations. Currently, magnetic resonance imaging (MRI) techniques are highly sensitive, and MRIs show iron accumulation in the nucleus of the pallidum, caudate and chiasmatic nuclei, and also in specific cortical region (van Duijn et al., 2017). In the neocortex and deep gray matter of the brain, AD patients had increased levels of iron than healthy controls, as shown in a subsequent study. Moreover, cognitive loss in AD sufferers was linked to changes in temporal lobe iron content with time (Damulina et al., 2020). When Aβ and tau interact, glial cells become hyperactive, a range of pro-inflammatory mediators are released, directing further inflammatory compounds toward the point of damage, leading to neural inflammation (Dhapola et al., 2021). It was shown that microglial iron overload reduces the secretion of insulin-degrading enzymes, which degrade Aβ and subsequently cause neurotoxicity by extracellular Aβ accumulation (Liu et al., 2022). The aberrant activation of microglia is caused by disruptions in brain iron equilibrium and neural ferroptosis, which contribute to enhanced neural inflammation. The release of inflammatory mediators by these improperly activated microglia exacerbates iron homeostasis disruptions and promotes neuronal inflammation (Wang M. et al., 2023). More studies has identified new ferroptosis symptoms in AD individuals, including elevated peroxidation of lipids and decreased systemic Xc-activity. Modifications in glutathione levels impact redox homeostasis and are linked with a higher risk of ferroptosis in AD individuals (Pocernich and Butterfield, 2012;
留言 (0)