
記住我




Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a genetic brain white matter disease with onset in infancy (van der Knaap et al., 1995a; Singhal et al., 1996). Compared to many other leukodystrophies, it has a mild clinical course. Almost all patients with MLC present with macrocephaly, which is obvious already in the first year of life (van der Knaap et al., 1995b). Brain MRI is characterized by diffuse signal abnormality and swelling of the cerebral white matter and the presence of cysts in subcortical areas, almost invariably in the anterior temporal lobe (Figures 1A–D) (van der Knaap et al., 1995a; van der Knaap et al., 1995b). Patients typically develop neurologic signs after a few years. Motor development is initially normal or slightly delayed, and later shows slow deterioration with ataxia and spasticity. Half of the patients lose the ability to walk without support and become wheelchair bound in their teens (Hamilton et al., 2018). Most MLC patients experience one or more seizures in their lifetime, and 63% of patients with classic MLC meet the criteria for clinical epilepsy (Hamilton et al., 2018). Seizures can typically be controlled with antiepileptic medication (Yalcinkaya et al., 2003; Dubey et al., 2018). Mild head trauma is often a trigger for seizures, and status epilepticus is more frequent in MLC than expected based on the mild epilepsy (Dubey et al., 2018). Behavioral and cognitive problems are common. The diagnosis of MLC is based on clinical and MRI criteria (van der Knaap et al., 2012; van der Knaap et al., 2018).
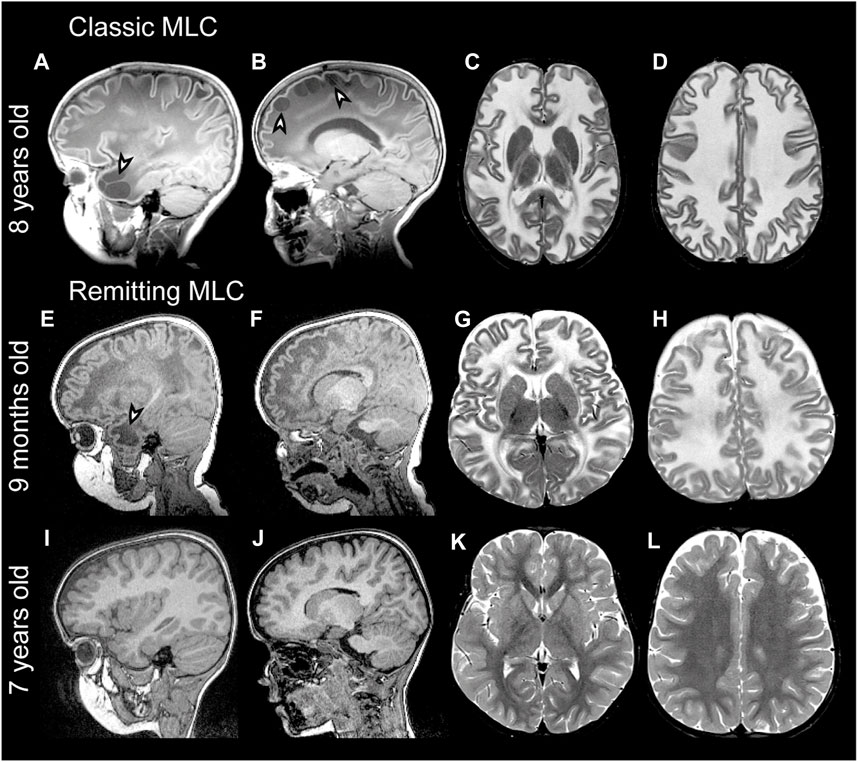
FIGURE 1. MRI findings in classic and remitting MLC patients. (A–D) MRI from an 11-year-old patient depicting an example of classic MLC. Anterior temporal and frontal subcortical cysts are visible in the sagittal T1-weighted MRIs in panels A and B (arrowheads). T2-weighted images in panel C and D show diffuse hyper intensity and swelling of the cerebral white matter with broadening of gyri (compare width of gyri in C and D to K and L). (E–H) MRI in a 9-month-old MLC patient and (I–L) images of the same patient at 7 years, showing the remitting phenotype. The anterior temporal cyst visible in panel E at 9 months (arrowhead) is no longer visible in panel I at 7 years. (F, J) No frontal subcortical cysts are present. Panels G, H, K, and L show that the cerebral white matter is initially T2-hyperintense and slightly swollen and that this T2-hyperintensity and swelling disappear over the years.
In 2001, the first gene linked to MLC was discovered and named MLC1 (Leegwater et al., 2001). Biallelic recessive MLC1 variants were found in many MLC patients. The associated disease is known as MLC1 (OMIM#604004). A remaining group of patients without MLC1 variants could be divided into patients with a classic clinical and MRI MLC phenotype and patients with initial signs of MLC followed by normalization of MRI and absence of motor and cognitive decline (van der Knaap et al., 2010) (Figures 1E-L). In 2011, a second MLC gene was discovered (Lopez-Hernandez et al., 2011a). This gene was initially called HEPACAM, but the name GLIALCAM is preferable because of its prominent expression in glial cells in the brain. Both patients with biallelic recessive GLIALCAM variants and patients with heterozygous dominant GLIALCAM variants were found. The small patient group with biallelic recessive variants in GLIALCAM has classic MLC and the associated disease is also known as MLC2A (OMIM# 613925). The larger group of patients heterozygous for a dominant GLIALCAM variant shows a remitting MLC phenotype, also known as MLC2B (OMIM# 613926). Macrocephaly and MRI properties are similar to classic MLC in the first year of life, but MRI greatly improves or normalizes in the following years and neurological regression does not occur (Lopez-Hernandez et al., 2011a). All patients with remitting MLC remain ambulatory, although some clumsiness can be present. In some patients head circumference also normalizes. Seizures and cognitive problems are less common in patients with remitting MLC, but autism is more common in these patients as compared to classic MLC patients (Lopez-Hernandez et al., 2011a; Hamilton et al., 2018).
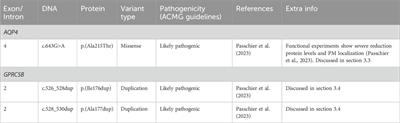
Recently two new genes were linked to MLC in the small group of patients that lack variants in MLC1 or GLIALCAM (Passchier et al., 2023). Heterozygous dominant variants in GPRC5B were found in patients with an MRI pattern and clinical course characteristic of classic MLC patients, and the associated disease is known as MLC3 (OMIM# 620447). A homozygous recessive variant in AQP4 was identified in two siblings with MLC typical of the remitting form of the disease, and this disease is known as MLC4 (OMIM# 620448).
The last comprehensive overview of genetic variants linked to MLC dates from 2006 (Boor et al., 2006). This was before the discovery of GLIALCAM, GPRC5B and AQP4 as additional genes linked to MLC. Since then, many new variants in all four MLC genes have been described in literature and new variants were discovered in the Amsterdam Leukodystrophy Center (ALC). In this study we provide an overview of all known variants in MLC1, GLIALCAM, GPRC5B and AQP4 that have been linked to MLC to date. We discuss particularly notable variants. We briefly highlight the link of MLC1 and GLIALCAM variants with psychiatric diseases, recapitulate what is known about MLC disease mechanisms from cellular, molecular and animal studies and provide an outlook for future research.
2 Materials and methodsWe made a list of known variants in MLC genes by performing an extensive literature search, supplemented with variants taken from the patient database of the ALC. We used the following accession numbers: NT_011526.7 and NM_015166.3 for MLC1, NT_033899.8 and NM_152722.4 for GLIALCAM (HEPACAM) NM_016235.3 for GPRC5B and NM_001650.7 for AQP4. All found variants were checked against the reference sequence, and nomenclature was updated, if necessary, making use of Alamut Visual version 2.9 (Interactive Biosoftware, Rouen, France). Interpretations of pathogenicity following ACMG guidelines were done for all variants (Richards et al., 2015).
2.1 Literature searchTo identify MLC1 and GLIALCAM variants described in the literature, we performed a PUBMED search using the search words ‘MLC1’, ‘GLIALCAM’, ‘HEPACAM’ ‘AQP4 MLC’ and ‘GPRC5B’. Articles published until July 2022 were included. We included papers that were written in English, Dutch or French. Only papers discussing patient data were included (e.g., descriptions of cloned plasmid variants without patient relevance were excluded). Variants were reported only when the coding sequence position was reported and when a conclusive MLC diagnosis (based on MRI) was reported in the study.
2.2 ALC diagnostic workflow and database inclusionPatients from the database of the ALC were included in this study upon conclusive MLC diagnosis by clinical features, MRI and genetic confirmation of variants in MLC1, GLIALCAM, GPRC5B or AQP4. Written informed consent was obtained from families for phenotyping.
The diagnostic workflow for MLC patients in the ALC is as follows: First, the diagnosis of MLC is established based on the presence of macrocephaly and characteristic MRI abnormalities now or in the past (Figure 1). Genetic testing starts with Sanger sequencing of MLC1. If no potentially pathogenic variants are found, this is followed by Sanger sequencing of GLIALCAM. When both are negative, and the MRI diagnosis is unambiguous, multiplex ligation-dependent probe amplification (MLPA) and cDNA analysis using lymphoblasts are performed for MLC1. If these do not uncover potentially pathogenic variants, next-generation sequencing (NGS, preferably whole genome sequencing (WGS)) is performed to identify potential rare (non-coding) variants.
2.3 Validation of variants impacting on MLC1 expressionThree reporter constructs were generated with the pNL1.1 vector (Promega), in which MLC1-expression regulating DNA sequences c.-2,645 to c.-1 (wild-type or with the c.-190A>G or c.-195T>C variant) were cloned directly upstream of the nanoluciferase open reading frame similarly as previously described (Hamilton et al., 2017). This DNA sequence includes the MLC1 core promoter and encodes the full 5′ untranslated region (5′UTR). Sanger sequencing was performed to confirm the MLC1 sequence with or without either of the two variants in the three p.NL1.1-MLC1 plasmids. Subsequently, U373 cells were cultured in DMEMF12 + 10% FBS. 24 h before transfection 3,000 cells were plated in white half area 96 well plates. The next day cells were co-transfected with a wild-type or mutant pNL1.1-MLC1 plasmid and the pGL3 plasmid (Promega) as internal standard using Fugene6 according to manufacturer’s instructions. The pGL3 plasmid encodes the firefly luciferase open reading frame under regulation of the SV40 promoter. Approximately 40 h after transfection, nanoluciferase and firefly luciferase activities were measured with a plate reader (Victor2; Perkin-Elmer Life Sciences, Waltham, MA), as described (Hamilton et al., 2017). The nanoluciferase signal was normalized to the firefly luciferase signal to obtain the relative expression driven by the wildtype and mutant MLC1 sequences. Statistical analysis was performed with Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test using GraphPad Prism 9 (GraphPad, USA). Statistically significant differences were defined as p ≤ 0.05. Data are represented as mean ± SEM.
2.4 Database submissionAll variants described in this study have been submitted to the LOVD database (www.lovd.nl): MLC1: https://databases.lovd.nl/shared/transcripts/00013671; HEPACAM: https://databases.lovd.nl/shared/transcripts/00009260; AQP4: https://databases.lovd.nl/shared/transcripts/00002726; GPRC5B: https://databases.lovd.nl/shared/transcripts/00008881.
2.5 UK biobankTo estimate allele frequency for some variants, data was obtained from approximately 500,000 participants from the UK Biobank (Bycroft et al., 2018), a population-based sample of adults in the UK with self-report surveys, linked electronic health records, and genotypic data. The National Research Ethics Service Committee North West–Haydock ethically approved this initiative (reference 11/NW/0382) and participants provided informed written consent. Data were accessed under application #16406.
3 Variants3.1 MLC1 variantsThe MLC1 gene is located on chromosome 22q13. The gene contains 12 exons and 11 introns (Figure 2A). The 5′UTR consists of exon 1 and part of exon 2. Predictions and experimental studies show that the MLC1 protein has 8 transmembrane regions with both the amino- and the carboxy-terminus residing in the cytoplasm (Figure 2B). MLC1 most likely forms a trimeric structure in the membrane (Hwang et al., 2021). The protein has very low homology with other proteins, with highest similarity (less than 20%) with the shaker-related voltage gated potassium channel Kv1.1 α-subunit (Teijido et al., 2004; Brignone et al., 2015). The exact function of MLC1 is not known. However, experimental studies have implicated an indirect role for MLC1 in cell ion and water homeostasis.
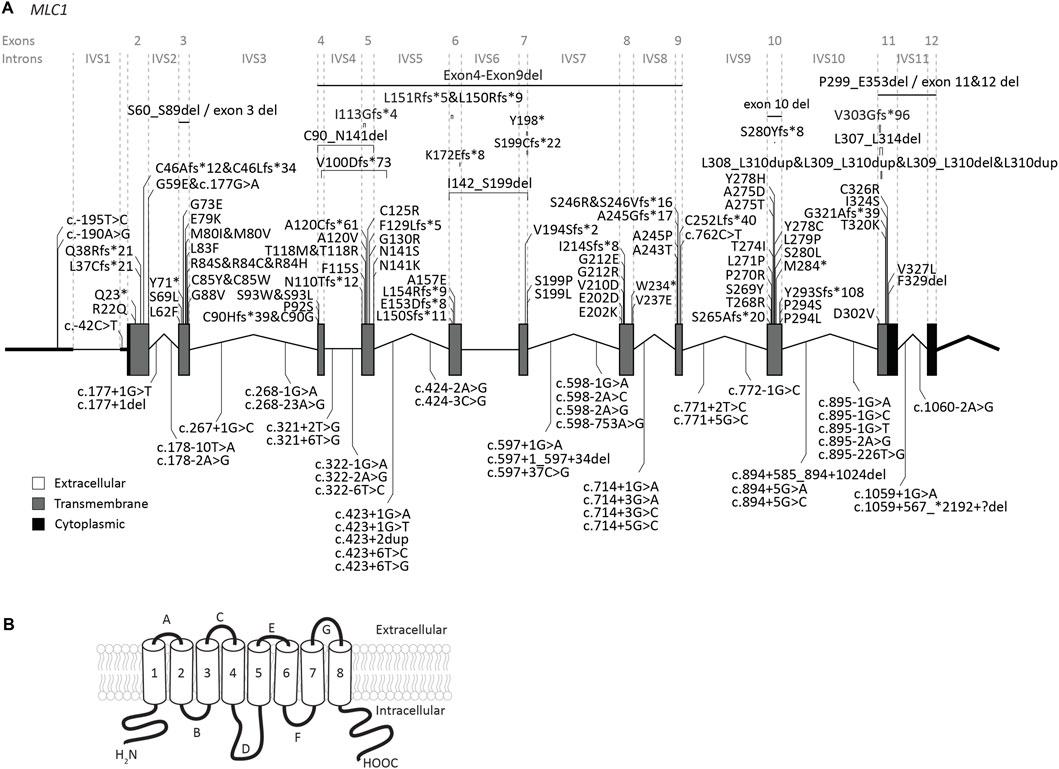
FIGURE 2. An overview of MLC1 variants found in MLC patients. (A) MLC1 is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above or below the gene schematic. For exonic variants the resulting peptide alterations are indicated, for intronic variants coding DNA alterations are indicated. Exonic variants are depicted in their relative positions. Intronic variants are depicted in their relative position roughly in the first or second half of the respective intronic region. (B) Schematic image of MLC1 in the cell membrane.
Our search yielded a total of 151 unique MLC1 variants in patients with a confirmed MLC diagnosis (Figure 2; Supplementary Table S1). Biochemical studies have been performed for some variants (see Supplementary Table S1) (Teijido et al., 2004; Duarri et al., 2008; Lopez-Hernandez et al., 2011b; Lanciotti et al., 2012; Capdevila-Nortes et al., 2013a; Sirisi et al., 2014; Xu et al., 2021). For most tested variants these studies reveal reduced plasma membrane levels, with retention of the protein in intracellular compartments (possibly the endoplasmic reticulum). In addition, protein stability is reduced for several variants. This might be a consequence of misfolding or defective oligomerization, leading to disrupted protein structure.
Several (possible) founder variants in MLC1 have been described. c.135dup; p.(Cys46Leufs*34) is a founder variant in East Indian individuals from the Agrawal community (Leegwater et al., 2002; Singhal et al., 2003; Gorospe et al., 2004). c.176G>A; p.(Gly59Glu) is a possible founder variant in Libyan Jews (Ben-Zeev et al., 2002). c.278C>T; p.(Ser93Leu) is common in Japanese individuals (Shimada et al., 2014), while c.824C>A; p.(Ala275Asp) is a founder variant accounting for the majority of MLC patients of Korean ancestry (Choi et al., 2017). c.908_918delinsGCA; p.(Val303Glyfs*96) is a founder variant in individuals with Egyptian ancestry.
3.1.1 Genotype-phenotype correlation for MLC1All MLC1 variants are recessive and cause classic MLC when present in homozygous or compound heterozygous form. It is known that clinical disease severity greatly varies between patients, and can even greatly differ for patients with the same MLC1 variants. For example, two siblings homozygous for the c.736A>C; p.(Ser246Arg) variant show a particularly mild phenotype, but still with considerable differences between them. No clear genotype-phenotype correlation has been established (Gorospe et al., 2004; Hamilton et al., 2018). Most patients with bi-allelic variants in MLC1 have slowly progressing disease. However, a low number of patients with bi-allelic MLC1 variants display radiological improvement in the course of years. Recently such a patient with radiological improvement was described (Mayayo-Vallverdú et al., 2023). The two variants in this patient (c.597+37C>G; p.? and c.895–1G>T; p.?) cause splicing defects and the researchers could detect a small amount of wild-type MLC1 transcript as well as wild-type MLC1 protein in peripheral blood leukocytes taken from the patient. Incomplete penetrance of the splice site variant could explain the residual MLC1 and might underlie the radiological improvement. Similarly, we observed radiological improvement for patients in the ALC database with variants upstream of the MLC1 open reading frame. These variants reduce protein expression (c.-195T>C; p.? and c.-190A>G; p.?; see description below in section 3.1.2). In these cases, a low level of residual wild-type MLC1 is also expected, which may explain the improvement. However, given the broad phenotypic spectrum of MLC patients and the rarity of patients affected by these specific variants, further studies are required to confirm whether low levels of residual wild-type MLC1 are indeed at the basis of radiological improvement.
3.1.2 Two variants upstream of the MLC1 open reading frame reduce expressionOur database contains three MLC patients, in whom Sanger sequencing revealed two variants of unknown significance in exon 1 of MLC1. One of these patients is heterozygous for the c.-190A>G; p.? variant, with the other MLC1 allele affected by another, known pathogenic variant. The second patient is heterozygous for the c.-195T>C; p.? variant, with the other MLC1 allele affected by a different known, likely pathogenic variant. The third patient is homozygous for the c.-195T>C; p.? variant. To assess if and how the c.-195T>C; p.? and the c.-190A>G; p.? variants affect MLC1 expression, a set of three reporter constructs was created, in which nanoluciferase expression was regulated by the wild-type or mutant sequences upstream of the MLC1 open reading frame. Both variants significantly reduce the expression of the downstream reporter by more than 50%, indicating that they are likely to reduce but not fully abrogate MLC1 expression in patients (Figure 3). In combination with the patients’ clinical and MRI phenotypes, we classified these variants as UV4, likely pathogenic.
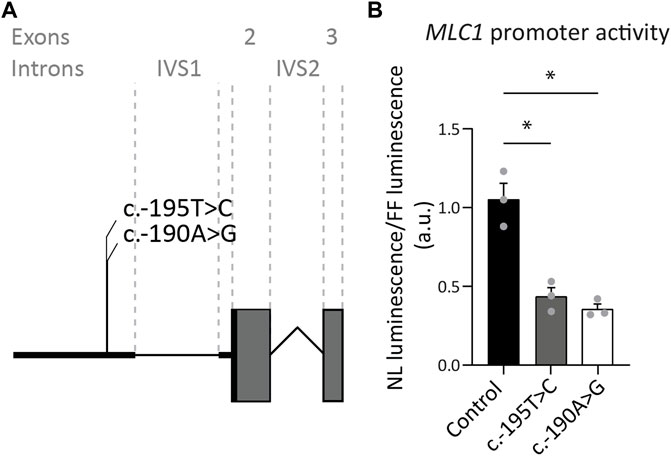
FIGURE 3. Exon 1 variants affecting expression of the downstream reporter. (A) Miniature schematic of 2 variants in the promoter region of MLC1. (B) Reporter gene assay depicting MLC1 promoter and 5′UTR activity in U373 cells. Readout was the ratio of Nano luciferase luminescence over Firefly luciferase luminescence in arbitrary units (a.u.). Experiments were performed in triplicate (n = 3), and each data point represents the average of 4 technical replicates. Graph shows means and individual data points of one experiment. Both the c.-190A>G and c.-195T>C variants significantly reduced expression of the downstream open reading frame of the reporter, reflected in a decrease in luminescence over fluorescence ratio. Brown-Forsythe ANOVA; F = 30.63 (2.000, 3.543) p = 0.0058. Dunnett’s T3 multiple comparisons test Control vs. c.-190A>G t = 6.576 df = 2.392 p = 0.0364. Control vs c.-195T>C t = 5.363 df = 3.085 p = 0.0222. *,p < 0.05.
3.1.3 A large intronic deletion in MLC1Our database contained a patient homozygous for the c.894+585_894+1024del; p.? variant in MLC1, detected by WGS, with clinical and MRI features of classic MLC and no variants found in GLIALCAM. Both parents were unaffected carriers. The variant causes a deletion of 440 nucleotides from intron 10 of MLC1. The variant is not listed in the 1,000 genomes database (www.1000genomes.org), and not found in the UK Biobank. This makes it unlikely that it represents a common polymorphism. The deletion reduces the number of GGGGGAUGGAGUCACUG repeats present in wild-type MLC1 RNA from 17 to 3. These repeats share similarity with previously described G-rich intronic splicing enhancers (Kralovicova and Vorechovsky, 2007). One possibility therefore is that the variant reduces intron 10 splicing. Alternatively, the deletion could lead to a reduction in RNA stability and thereby reduce MLC1 protein expression. Therefore, we classify the c.894+585_894+1024del; p.? variant as UV3 (variant of unknown significance).
3.1.4 MLC1 variants that affect splicing41 variants in MLC1 affect splicing. Most of these are in canonical splice sites. For variants outside of canonical splice sites, including some deep intronic variants (Mancini et al., 2012) a splicing defect was confirmed using cDNA analysis. The c.597+37C>G; p.? variant discussed in section 3.1.1 (Mayayo-Vallverdú et al., 2023), creates a splice acceptor site in intron 7. cDNA analysis shows that the variant affects RNA splicing and leads to skipping of exon 7 and partial retention of intron 7.
3.1.5 (Likely) benign variants in MLC1 and variants with an uncertain link to MLCSeveral likely benign MLC1 variants have been reported (Leegwater et al., 2002; Shukla et al., 2011; Wang et al., 2011), and one was found in the ALC database. These are listed in Table 1, together with their allele frequency in the UK Biobank. For some variants in MLC patients reported in literature it is not clear whether they cause disease. These variants are listed in Table 1. The variant c.858C>G; p.(Ile286Met) was observed on the paternal allele in only one individual that carried another known pathogenic variant on the same allele (Cao et al., 2016). The authors describe that this patient has classic MLC. No other patients with this variant have been found. The variant is not found in the gnomAD database or in the UK Biobank, a large UK cohort (Table 1). Based on this information it is not possible to conclude whether the variant is disease causing. An individual with MLC with a heterozygous c.95C>T; p.(Ala32Val) variant on the maternal allele was described by Wang and others (Wang et al., 2011). No MLC1 variant was found on the paternal allele. At the time of this study MLC1 was the only known MLC gene. It is therefore possible that this individual had variants in another MLC gene, or that additional hard to detect MLC1 variants were missed. In a follow-up study from the same team, Cao and others (Cao et al., 2016) describe this variant in a patient who also has a dominant variant in GLIALCAM. This could be the same patient as described in the earlier study. A follow-up MRI for this patient was not described, making it impossible to assess whether this patient had a remitting phenotype. The c.95C>T; p.(Ala32Val) MLC1 variant has an allele frequency of 2.02*10^-5 and an allele count of 19 in the UK Biobank (Table 1).
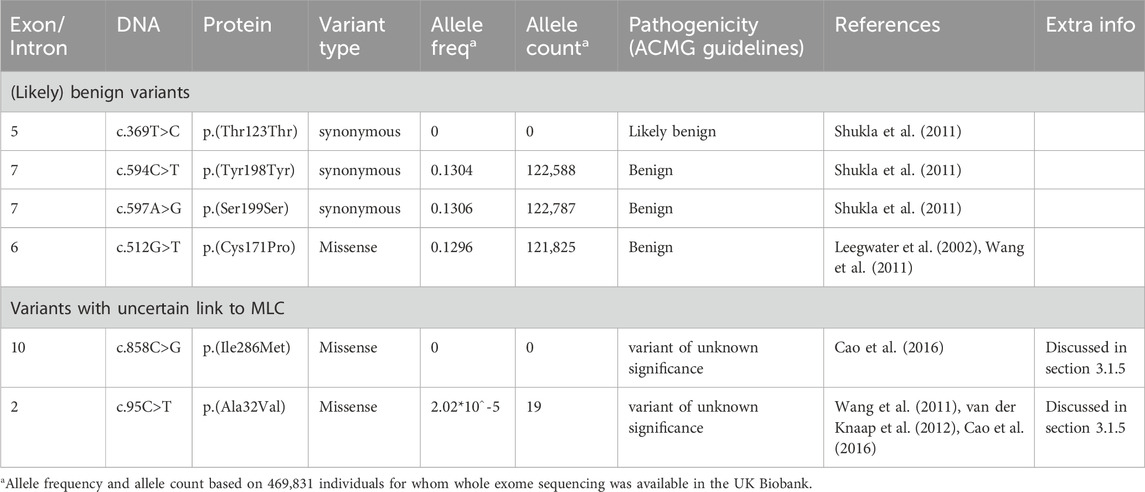
TABLE 1. (Likely) benign variants in MLC1 and variants with an uncertain link to MLC.
3.2 GLIALCAM variantsThe GLIALCAM gene is located on chromosome 11q24. It contains 7 exons and 6 introns (Figure 4A). The protein GlialCAM, encoded by GLIALCAM, is an immunoglobulin-like transmembrane adhesion protein of 417 amino acids. It contains two N-terminal immunoglobulin domains (IgV and IgC2), a transmembrane domain and an intracellular C-terminal domain (Capdevila-Nortes et al., 2015) (Figure 4B). GlialCAM tightly interacts with MLC1 (Capdevila-Nortes et al., 2013b), acts as an auxiliary subunit for ClC-2 chloride channels (Jeworutzki et al., 2012) and regulates Connexin-43 mediated gap-junctional coupling (Wu et al., 2016). Before GLIALCAM was linked to MLC, it was mainly known as a cancer gene (Moh et al., 2008; He et al., 2010; Zhang et al., 2011). A total of 29 unique variants in GLIALCAM were identified in patients with either classic or remitting MLC (Figure 4; Table 2).
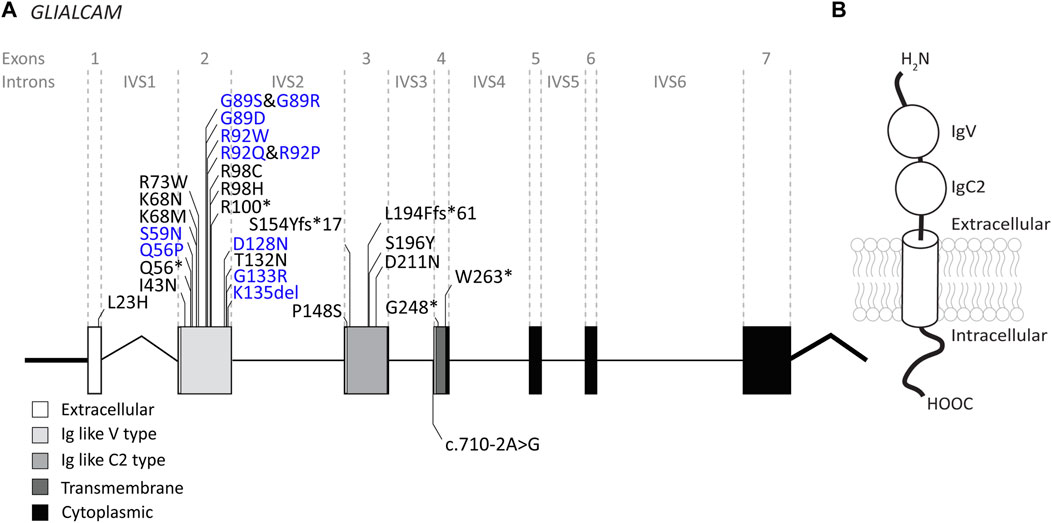
FIGURE 4. An overview of GLIALCAM variants found in MLC patients. (A) The GLIALCAM gene is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above or below the gene schematic. Resulting peptide alterations are indicated for exonic variants, and coding DNA alteration for an intronic variant. Variants are depicted in their relative positions. Dominant GLIALCAM variants are depicted in blue; recessive variants are depicted in black. (B) Schematic representation of GlialCAM in a cell membrane.
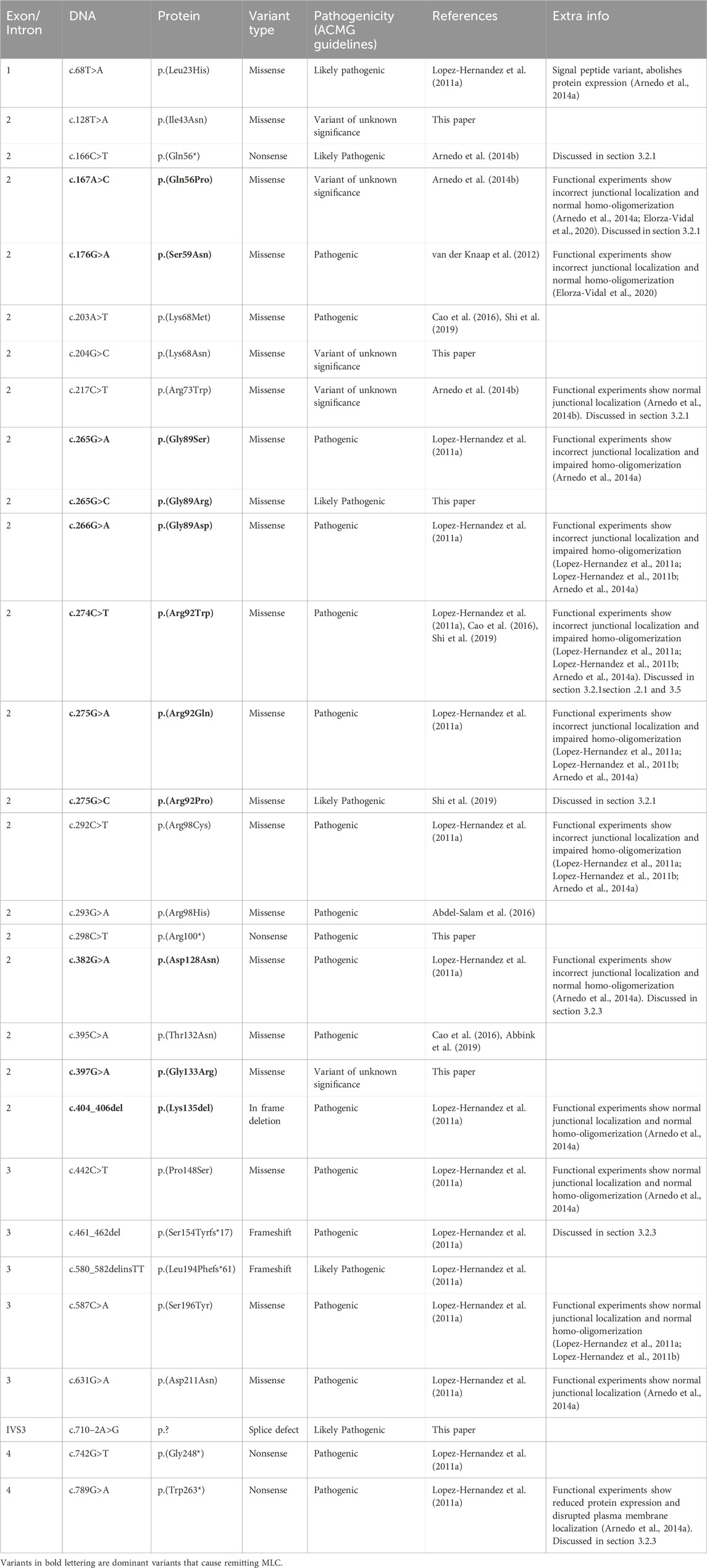
TABLE 2. GLIALCAM variants found in MLC patients.
3.2.1 Genotype-phenotype correlation: dominant GLIALCAM variants cause remitting MLCFor GLIALCAM variants, there is a clear genotype-phenotype correlation: Dominant variants lead to remitting MLC when present in heterozygous form. Variants classified as recessive lead to classic MLC when present in homozygous or compound heterozygous form. Some patients from the ALC database have a dominant variant on one allele and a recessive variant on the second allele. These patients have classic MLC.
Of the 29 GLIALCAM variants found in MLC patients, 11 have been reported to have a dominant effect (Figure 4; depicted in blue; bold in Table 2). All of these variants are located in exon 2. In addition, with the exception of one amino acid deletion, all dominant variants are missense. Heterozygous presence of these variants leads to remitting MLC. For some variants (c.167A>C; p.(Gln56Pro), c.274C>T; p.(Arg92Trp) and c.275G>C; p.(Arg92Pro)) it was reported that family members carried these variants but were not diagnosed with MLC in their youth. This indicates either reduced penetrance of the variants (meaning that some individuals with a dominant variant do not have a disease), or it means that the remitting MLC phenotype can be so mild in some individuals that it remains undiagnosed.
Functional experiments on recessive and dominant GLIALCAM variants show that most variants disrupt localization of GlialCAM to cell-cell junctions (Lopez-Hernandez et al., 2011a; Lopez-Hernandez et al., 2011b; Arnedo et al., 2014a). Structurally, all dominant variants affect the first extracellular immunoglobulin domain of the GlialCAM protein, while recessive variants are found in parts of the gene encoding the extracellular or transmembrane parts of the protein. Biochemical experiments suggest that dominant variants disrupt homophilic GlialCAM-GlialCAM interactions; possibly more specifically the interactions in trans with GlialCAM on neighbouring cells (Elorza-Vidal et al., 2020).
We report the c.217C>T; p.(Arg73Trp) and c.166C>T; p.(Gln56*) variants as recessive, because they have only been found in compound heterozygous form in one patient (Arnedo et al., 2014b). However, there is marked improvement of brain MRI abnormalities and mild clinical symptoms, more consistent with remitting MLC. c.166C>T; p.(Gln56*) is predicted to produce no functional protein. No clear effect of the c.217C>T; p.(Arg73Trp) variant was observed in cellular and biochemical assays. One possibility is that the full loss of function caused by the c.166C>T; p.(Gln56*) variant by itself is sufficient to cause a remitting MLC phenotype (see also the discussion on GLIALCAM hemizygosity below). Alternatively, the c.217C>T; p.(Arg73Trp) variant might act in a dominant fashion. To fully understand the consequence of these two variants therefore requires either a better mechanistic understanding of GlialCAM function, or the observation of novel patients with only one of these variants.
3.2.2 GLIALCAM hemizygosity in Jacobsen syndromePartial deletion of the terminal part of chromosome 11q leads to Jacobsen syndrome (Mattina et al., 2009). MRI abnormalities resembling MLC have been described in Jacobsen syndrome patients already long ago, which led to the speculation that a leukodystrophy gene could be located in this region (Wardinsky et al., 1990; Gutmann, 1991). Indeed, deletion of 11q24 including GLIALCAM was later linked to MLC-like white matter abnormalities with diffuse signal abnormality and swelling of the cerebral white matter in several patients (Yamamoto et al., 2015; Patel et al., 2019; Wolf and van der Knaap, 2020). Importantly, the MRI phenotype in Jacobsen syndrome is remitting (similar to what is seen in remitting MLC; see Figure 5 for an example from the ALC database). Since MRI abnormalities are not observed in all Jacobsen patients, either penetrance of such abnormalities upon GLIALCAM hemizygosity is not complete, or white matter abnormalities were missed because they resolved before the first MRI (Ono et al., 1996). This suggests that hemizygosity for GLIALCAM may lead to a clinical phenotype similar to remitting MLC.
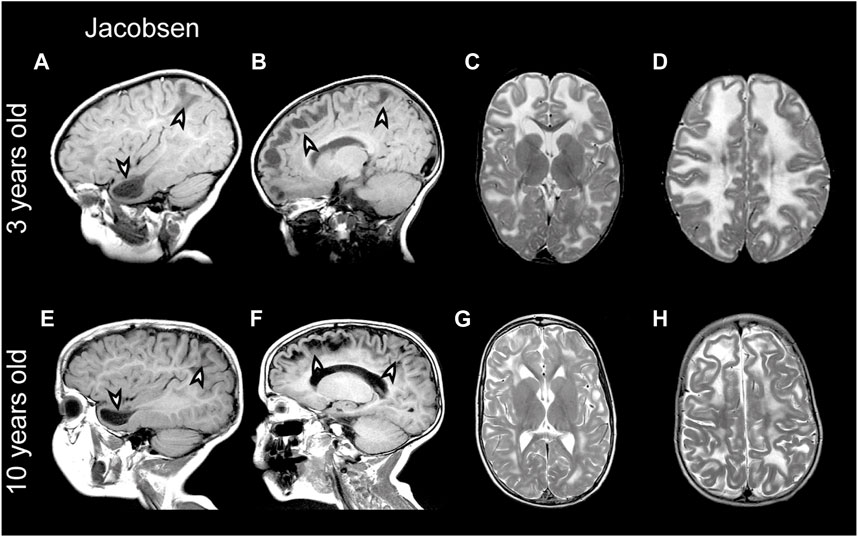
FIGURE 5. MRI findings in a Jacobsen syndrome patient. (A–D) MRIs from a 3-year-old Jacobsen syndrome patient with a chromosomal deletion that includes GLIALCAM. Numerous anterior temporal and frontal subcortical cysts are visible in the sagittal T1-weighted MRIs in panels A and B (arrowheads). T2-weighted images in (C and D) reveal extensive signal abnormality and swelling of the cerebral white matter. (E–H) Follow-up of the same patient at 10 years of age. Subcortical cysts persist [arrowheads in (E,F)], but there is clear improvement in signal abnormality and swelling of the cerebral white matter.
3.2.3 (Likely) benign variants in GLIALCAM and variants with an uncertain link to MLCFor some variants it is not possible to establish their link to MLC. c.461_462del; p.(Ser154Tyrfs*17) (Table 2) and c.789G>A; p.(Trp263*) (Table 2) were found in one patient on the same allele. It is likely that the first variant, which causes a premature stop codon, already disrupts the expression of the full-length GlialCAM protein. Still, because the second variant also causes a premature stop codon, we have classified both variants as pathogenic.
c.862C>T; p.(Arg288Cys) was found in an individual with remitting MLC. This individual had the c.382G>A; p.(Asp128Asn), a known dominant variant, on the same allele (Lopez-Hernandez et al., 2011a). The allele count of c.862C>T; p.(Arg288Cys) in the UK Biobank is 122 (heterozygous only) with an allele frequency of 1.29*10−4 and in the gnomAD database the allele count is 156 with one occurrence in homozygous state (Table 3). Based on this information it is not possible to say whether the variant is pathogenic. We classify this variant as a variant of unknown significance.

TABLE 3. (Likely) benign variants in GLIALCAM and variants with an uncertain link to MLC.
Certain dominant GLIALCAM variants have been observed in compound heterozygous state with an intronic variant of unknown consequence (c.877+101G>T; p.?). This variant was also found in two patients with additional biallelic GLIALCAM variants. The allele frequency of this variant in the UK biobank, gnomAD and 1000genomes (3.43%) database is high (Table 3). We conclude that this variant is benign. c.582C>T; p.(Leu194Leu) was found on one allele together with a known pathogenic variant. The variant is synonymous and not predicted to affect splicing. Allele frequency in the UK Biobank is low. We conclude that this variant is likely benign.
3.3 AQP4 variantsThe AQP4 gene is located on chromosome 18q11.2 and contains 5 exons and 4 introns (Figure 6A). It encodes the water channel aquaporin-4 (AQP4), the most abundant aquaporin in the brain (King et al., 2004). AQP4 has two main isoforms due to the use of two different translation initiation sites (Neely et al., 1999; Verkman et al., 2011): the longer M1 isoform and a shorter M23 isoform. Both isoforms, among other isoforms, are expressed in astrocytes and assemble as heterotetramers (Jorgacevski et al., 2020). AQP4 has 6 transmembrane domains and 2 highly conserved NPA domains that form the pore in the membrane (Ho et al., 2009) (Figure 6B). Both the C- and the N-terminus reside intracellularly. AQP4 is of vital importance for brain ion and water homeostasis (Nagelhus and Ottersen, 2013). It shares its location in astrocyte endfeet with MLC1 and GlialCAM (Nagelhus et al., 2004).
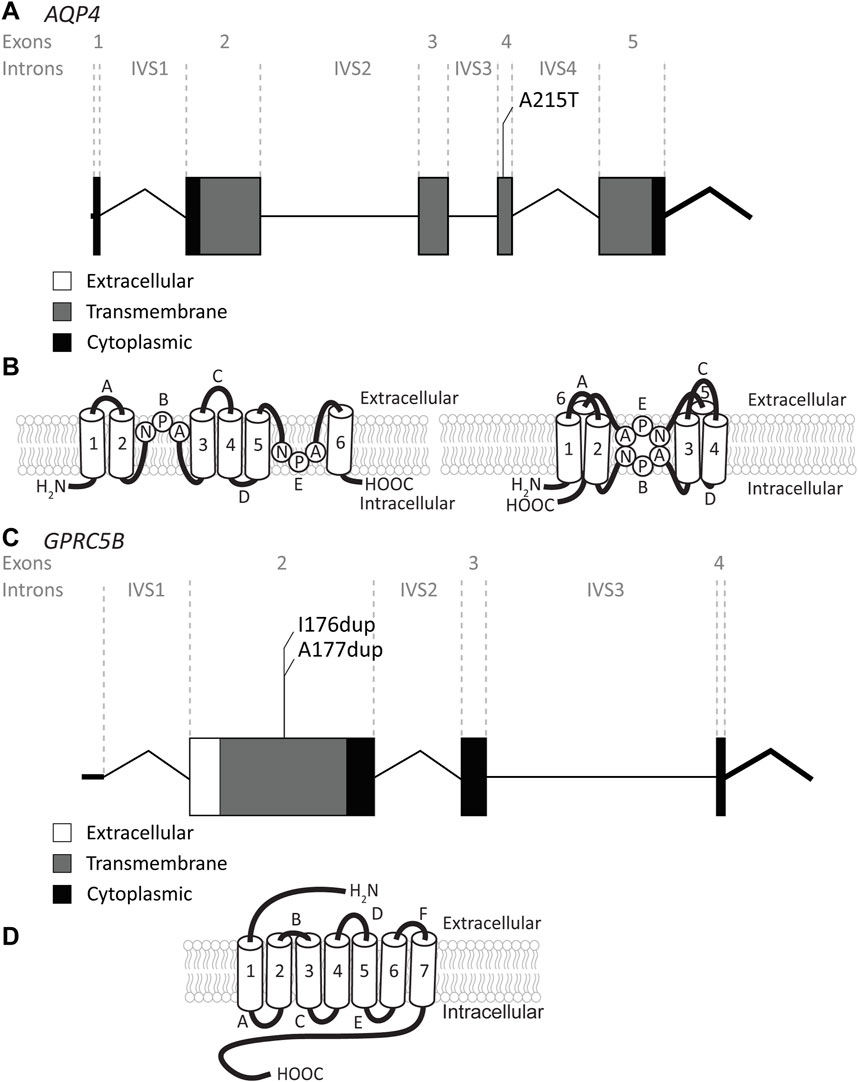
FIGURE 6. An overview of AQP4 and GPRC5B variants found in MLC patients. (A) AQP4 is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above the gene schematic. The resulting peptide alteration is in its relative position. (B) A schematic representation of AQP4 in the membrane. Amino acids of the NPA motif are depicted. (C) As in (A) however here, GPRC5B is depicted with its two peptide alterations. (D) A schematic representation of GPRC5B in the membrane.
Recently, a recessive variant in AQP4 was found to lead to remitting MLC (Passchier et al., 2023). This is the first variant found in AQP4 to be disease linked and offers new insight into MLC disease mechanisms. The variant affects the key pore forming NPA motif in the AQP4 protein, exchanging an alanine, which is a hydrophobic amino acid, for a threonine, a hydrophilic amino acid (c.643G>A; p.(Ala215Thr) Figure 6A; Table 4). This leads to a loss of AQP4 at the cell membrane and therefore, a loss of function of the protein. Why an AQP4 defect would lead to remitting MLC is unknown. Passchier et al. describe that under certain conditions (e.g., massive overexpression) mutant AQP4 retains some function (Passchier et al., 2023). The physiological relevance of this is unclear, but this might form a basis for the observed radiological improvement. An alternative explanation could be redundancy in brain ion and water homeostasis, potentially with other aquaporins taking over the role of AQP4.
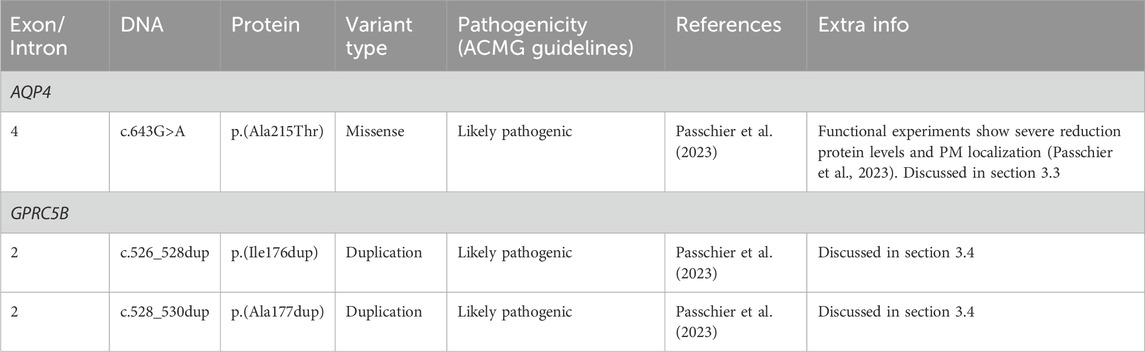
TABLE 4. AQP4 and GPRC5B variants found in MLC patients.
3.4 GPRC5B variantsThe GPRC5B gene is located on chromosome 16p12.3 and contains 4 exons and 3 introns. Exon 1 encodes the 5′UTR (Figure 6C). GPRC5B encodes the orphan G protein-coupled receptor (GPCR) GPRC5B. It is a class C GPCR and has 7 transmembrane regions and a relatively short extracellular N terminus compared to other GPCRs of the same class (Figure 6D). Multiple GPRC5B isoforms are known, including a long and a short isoform, as well as a brain specific isoform (Cool et al., 2010). GPRC5B is expressed in various tissues and cell types, including adipocytes, pancreatic islets, kidney podocytes and vascular smooth muscle cells. The receptor modulates inflammatory signalling through the NF-κB pathway, and has been linked to various disease states including diabetes (Soni et al., 2013), kidney disease (Zambrano et al., 2019) and artherosclerosis (Carvalho et al., 2020). Few studies have looked at GPRC5B function in the brain (Kurabayashi et al., 2013; Sano et al., 2018), but recently GPRC5B was identified as an MLC1/GlialCAM interacting protein that is expressed in astrocyte endfeet (Alonso-Gardon et al., 2021). Two dominant de novo variants in the fourth transmembrane region of GPRC5B were recently described in patients with classic MLC that had no variants in MLC1, GLIALCAM or AQP4 (Passchier et al., 2023). These two variants are specific amino acid duplications of two neighbouring amino acids in the fourth transmembrane domain of the GPCR (c.526_528dup; p.(Ile176dup) and c. 528_530dup; p.(Ala177dup); Figure 6C; Table 4). How these variants affect the function of GPRC5B is not yet understood.
3.5 Psychiatric and neurodevelopmental disorders linked with MLC1 and GLIALCAM variantsAbnormalities of the brain white matter have been consistently associated with psychiatric disorders (Fields, 2008). Psychiatric symptoms are therefore often seen in leukodystrophies, even sometimes as presenting symptoms (Costei et al., 2021). A clear example is the frequent occurrence of psychosis in Metachromatic Leukodystrophy (MLD OMIM# 250100) (Hyde et al., 1992).
Regarding neurodevelopmental disorders, autistic features are common within the population of MLC patients (Lopez-Hernandez et al., 2011a). Surprisingly, autistic features are seen in ∼25% of patients with remitting MLC caused by dominant GLIALCAM variants, while this is only 9% for patients with classic MLC (Hamilton et al., 2018). Therefore, while motor symptoms are invariably milder in remitting MLC, the occurrence of autistic features is higher. The basis for this difference is not understood. It suggests that dominant GLIALCAM variants disrupt a function of GlialCAM that is separate from its role in astrocyte endfeet. For example, a recent study showed that astroglial release of GlialCAM from exosomes regulates neuronal axon outgrowth and dendritic spine formation (Jin et al., 2023). Both of these processes have been implicated in the etiology of autism (Gilbert and Man, 2017). While speculative, such a specialized function of GlialCAM might explain why dominant GLIALCAM variants more often lead to autistic features.
Genetic screens in cohorts of patients with autism spectrum disorder (ASD) uncovered additional heterozygous missense variants in GLIALCAM. The c.274C>T; p.(Arg92Trp) variant, which was previously found in a remitting MLC patient (Lopez-Hernandez et al., 2011a), was independently discovered in a study on ASD patients (Iossifov et al., 2014). The c.437C>T; p.(Ser146Leu) variant was discovered in another study on ASD patients (Li et al., 2017). Another new GLIALCAM variant that has not been observed in MLC patients, c.505T>C; p.(Ser169Pro), was found in a study on patients with intellectual disability (Lelieveld et al., 2016). Finally, a splice site variant in GLIALCAM, c.803+1G>A; p.? was found in an ASD cohort. This variant was classified as variant of unknown significance (Zhou et al., 2019). Together, these studies further substantiate the link between specific dominant GLIALCAM variants, ASD and intellectual disability.
For MLC1 it has been suggested that specific variants in heterozygous state are linked to schizophrenia or bipolar affective disorder, two psychiatric disorders thought to share a common etiology. A rare missense variant in MLC1 (c.1121C>A; p.(Leu309Met)) was associated with periodic catatonia, a familial subtype of catatonic schizophrenia (#OMIM605419) in a large pedigree (Meyer et al., 2001), although this variant was not found in other cohorts of schizophrenia or bipolar affective disorder patients (Meyer et al., 2001). Additional MLC1 variants have been significantly associated with schizophrenia and bipolar affective disorder in an Indian cohort (Devaney et al., 2002; Ewald and Lundorf, 2002; Jorgensen et al., 2002; McQuillin et al., 2002; Rubie et al., 2003; Kaganovich et al., 2004), with confirmation of two intronic MLC1 variants in an independent study, where they were specifically associated with periodic catatonia and not with other types of schizophrenia (Selch et al., 2007). Interestingly, variants linked to bipolar disorder or schizophrenia have not been observed in MLC patients. In addition, the c.1121C>A; p.(Leu309Met) variant has no influence on MLC1 protein expression levels in cellular studies (Teijido et al., 2004). Therefore, while these findings
留言 (0)