
記住我
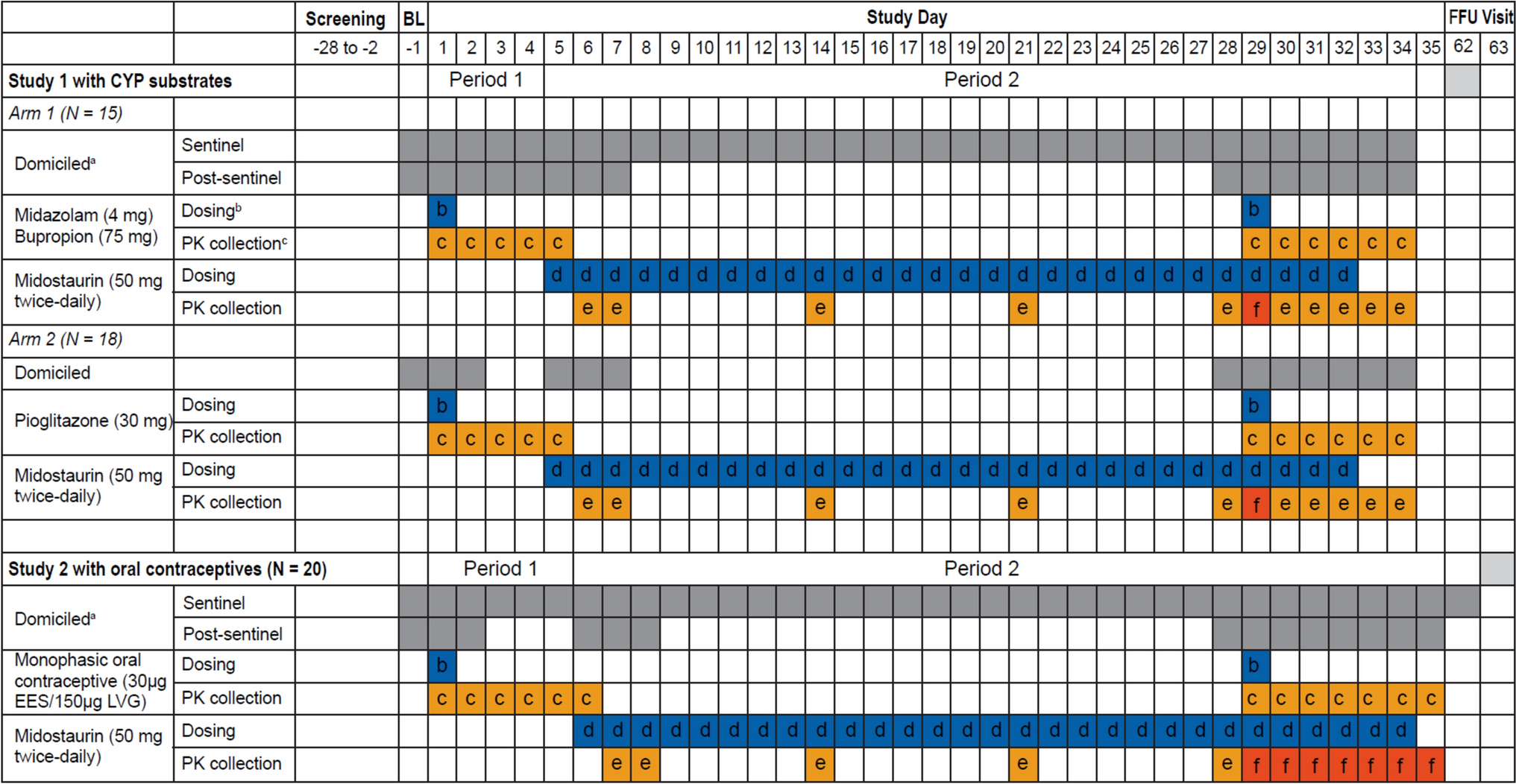
Study 1 with CYP substrates was a Phase 1, open-label, fixed-sequence DDI study in healthy adult participants. Participants were screened for eligibility and then admitted to the study center for baseline evaluation. There were two treatment arms—participants in Arm 1 received a single oral dose of 4 mg midazolam, 75 mg bupropion, and twice daily 50 mg midostaurin for 28 days; participants in Arm 2 received single oral dose of 30 mg pioglitazone and twice daily 50 mg midostaurin for 28 days. There were two treatment periods in each arm (Fig. 1).
Fig. 1
Study 1 with CYP substrates and Study 2 with oral contraceptives. BL baseline, D day, EES ethinylestradiol, FFU final follow-up, LVG levonorgestrel, OC oral contraceptive, PK pharmacokinetic. aFor Arm 1 only of Study 1: The participants were domiciled for the entire Period 1 and 2 (day − 1 to day 34) during sentinel dosing. Post-sentinel participants were domiciled from day − 1 to day 7 after morning dose of midostaurin and returned to the study center in the morning on day 8 to day 27 to take their morning dose of midostaurin supervised and were provided their evening dose to take at home. For Study 2: Participants were domiciled for the entire Period 1 and 2 during sentinel dosing (day − 1 to day 35). Post-sentinel subjects were domiciled from day − 1 to day 2, day 6 to day 8, and day 28 to day 34 of Period 1 and Period 2. They were provided their morning and evening dose of midostaurin supervised from days 28 to 33. bSingle dose. cFull profile up to 48 h (midazolam), 96 h (bupropion and pioglitazone), and 120 h (monophasic oral contraceptives). dTwice daily. eTrough concentrations on days 7, 8, 14, 21, and 28 (in the morning). fPK profile
During treatment Period 1 of each arm, a single oral dose of the cocktail drug substrates (midazolam and bupropion for Arm 1 and pioglitazone for Arm 2) was administered on day 1, and plasma concentrations of the drug substrate(s) and their respective metabolites were followed up to the morning of day 3 for midazolam (Arm 1) and until the morning of day 5 for bupropion (Arm 1) and pioglitazone (Arm 2). Treatment Period 1 was immediately followed by treatment Period 2. During treatment Period 2, participants in both Arms 1 and 2, received continuous treatment with midostaurin at 50 mg twice daily for 28 days (from day 5 to day 32). On day 29, participants received a single dose of the cocktail drug substrates (midazolam and bupropion for Arm 1 and pioglitazone for Arm 2) along with their morning dose of midostaurin. Plasma concentrations for midostaurin, its metabolites (CGP52421 and CGP62221), and the different drug substrates and their metabolites were then collected for 96 h until the morning of day 33. The participants continued to receive midostaurin until the evening of day 32, after which a final follow-up visit occurred 30 days after the last dose administration (day 62) for safety assessment.
Study 2 with oral contraceptives was a single-arm Phase 1, open-label, fixed-sequence DDI study in healthy female participants with no childbearing potential (Fig. 1). During Period 1, participants received a single dose of the oral contraceptive composed of 150 μg LVG and 30 μg EES and plasma concentrations were followed for 5 days. This period was immediately followed by Period 2. In Period 2, the participants received continuous treatment with midostaurin at 50 mg twice daily for 28 days (from day 6 to day 33). On day 29, the participants received a single dose of the oral contraceptive together with their morning dose of midostaurin.
PK samplingSequential PK sampling was conducted according to the assessment schedule for both studies. For Study 1 with CYP substrates, PK samples were collected at specified time points until 48 h post-dose for midazolam (pre-dose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 h) and until 96 h for bupropion and pioglitazone (pre-dose, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, and 96 h). For both Arm 1 and Arm 2, midostaurin trough samples were collected prior to morning doses on days 6, 7, 14, 21, 28, 30, 31, 32 and 33; and the midostaurin PK profile on day 29 at 0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h post-dose.
For Study 2 with oral contraceptives, trough PK samples were collected at pre-dose in the morning on days 7, 8, 14, 21, 28, 30, 31, 32, 33, and 34. PK samples for EES and LVG were collected on day 1 at pre-dose (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, and 120 h post-dose. Serial samples for EES and LVG were collected on day 29 at pre-dose (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24 (day 30), 48 (day 31), 72 (day 32), 96 (day 33), and 120 (day 34) hours post-dose. Midostaurin PK sampling was similar to that in Study 1.
Sentinel dosingBoth studies implemented a sentinel dosing approach to ensure the safety of the healthy participants receiving 50 mg twice daily midostaurin for 28 days (Fig. 1). The study initially enrolled three participants in the sentinel dosing cohort. These three participants were domiciled for the entire Period 1 and Period 2 of sentinel dosing. After the last dose of midostaurin, subjects remained in the study center for additional safety observation until 36 h post-dose, and a complete safety assessment was performed prior to discharge.
The study used specific safety criteria for guiding further enrollment, stopping the study, and/or progressing to post-sentinel dosing, including leukopenia or febrile neutropenia of Common Terminology Criteria for Adverse Events (CTCAE) Grade ≥ 3, aspartate aminotransferase (AST)/alanine aminotransferase (ALT) ≥ 3 times the upper limit of normal, or bilirubin ≥ 2 times the upper limit of normal. Detailed list is provided in the Online Resource 1.
Post-sentinel dosingIn Study 1 with CYP substrates, participants in Arm 1 were domiciled at the study center from day − 1 to day 7 (after third morning dose of midostaurin); and participants in Arm 2 were domiciled from day − 1 to day 2 and again from day 5 to day 7 (after third morning dose of midostaurin). During the out-participant period, they returned to the center for morning midostaurin dosing and received their evening dose to take at home. Safety checks were performed weekly. In Period 2, participants were domiciled from day 28 to day 33 (Arm 1 and Arm 2) and received a safety assessment prior to discharge. In Study 2 with oral contraceptives, participants were domiciled from day − 1 to day 2 of Period 1 and the first 2 days of Period 2 (day 6 to day 8) for PK sampling, dosing, and safety assessments. During the out-participant period, they returned to the center for morning midostaurin dosing and received their evening dose to take at home. In Period 2, participants were domiciled from day 28 to day 34 and received a safety assessment prior to discharge. A final follow-up visit for a safety assessment occurred 30 days after the last dose administration, and the total study duration ranged from approximately 90 to 91 days.
Study participantsStudy 1 with CYP substrates enrolled healthy adult male and female participants of non-childbearing potential, aged between 18 and 65 years (inclusive) with a body mass index (BMI) between 18.0 and 29.9 kg/m2, and with no history of cardiac disease or significant electrocardiogram (ECG) abnormalities. Key exclusion criteria included a history of cardiac disease, history of prolonged QT-interval syndrome or cardiac disease, and contraindication or hypersensitivity to any drug or metabolites from a similar class as the study drug. Study 2 with oral contraceptives enrolled healthy female participants aged 18–65 years (inclusive), with non-childbearing potential, with a BMI between 18.0 and 29.9 kg/m2, and with no history of arterial or venous thromboembolism. Participants were excluded if they were allergic to the study drug or its metabolic components or had a history of ECG abnormalities that included prolonged QT-interval syndrome or cardiac disease. Detailed inclusion and exclusion criteria for both the studies are available in Online Resource 2.
Outcome measuresThe primary endpoint for Study 1 with CYP substrates and Study 2 with oral contraceptives was the derived PK parameters (non-compartmental analysis [NCA]) of a single dose of midazolam, bupropion, pioglitazone, and EES and LVG when administered alone and concomitantly with midostaurin, respectively. The primary parameters measured were Cmax, AUClast, and AUCinf, while secondary parameters included Tmax, CL/F, Vz/F, and T1/2. Definitions for the measured PK parameters are provided in Online Resource 3. The secondary endpoints for both the studies were adverse events (AEs) based on the CTCAE grade (severity) and frequency, as well as other safety data such as (ECG) and laboratory parameters. The other secondary endpoint was midostaurin (multiple dose) and metabolite (CGP52421 and CGP62221) PK parameters Cmax,ss, AUCtau interval tau at steady-state, Tmax,ss, CL/F, and Vz/F. The exploratory endpoint was PK parameters (NCA) of the drug substrate’s metabolites when administered alone and concomitantly with midostaurin, including Cmax, Tmax, AUClast, and metabolic ratios (Online Resource 3).
Statistical analysisThere were three population sets, the full analysis set (FAS), the safety analysis, set and the PK analysis sets (PASs). FAS and safety analysis set were identical in the studies. The PAS included all participants who provided an evaluable PK profile for at least one period. In Study 1, there were three PASs, one for probe drugs in each arm: midazolam and bupropion in Arm 1 (PAS1), pioglitazone in Arm 2 (PAS2), and one for midostaurin (PAS3). In Study 2, there were two PASs, one for oral contraceptive and the other for midostaurin.
Analysis of the primary PK parameters to estimate the effect of midostaurin (and its metabolites CGP62221 and CGP52421) on the PK of midazolam (and its metabolite 1ʹ-hydroxy midazolam), bupropion (and its metabolite hydroxy-bupropion), pioglitazone (and its metabolite hydroxy-pioglitazone), and LVG and EES components of the oral contraceptives were done using a linear mixed model.
Geometric mean ratios of the PK parameters obtained in the treatment with the drug substrate + midostaurin were compared to those obtained in treatment with the drug substrate alone. Point estimates and two-sided 90% confidence interval (CI) for the difference between means of test and reference treatment (test-reference) were calculated, and were anti-log transformed to obtain geometric mean ratios. The median and the range of differences in the Tmax values of midazolam, bupropion, and pioglitazone with 90% CI were calculated and presented for test versus reference.
Safety data were analyzed by number and percentage of participants with AEs and tabulated by system organ class (SOC) and preferred term (PT).
Physiologically based pharmacokinetic modelA PBPK model using Simcyp (Certara, L.P., Simcyp, Sheffield, UK) had been previously developed and verified to model the PK of midostaurin and its metabolites (CGP52421, and CGP6221) after 50 mg twice daily dosing in healthy volunteers [4]. The Sim-Healthy Volunteer population in Simcyp was used for further model refinement and qualification for PK and DDI predictions of midostaurin with midazolam using clinical data (Study 1 with CYP substrates) in the current analysis [15]. The model was applied to predict DDI with midazolam at high dose of midostaurin (100 mg twice daily). Additionally, the PBPK model was also used to qualify the DDI prediction of midostaurin (50 mg twice daily) with an oral contraceptive, eg., EES from clinical data (Study 2 with oral contraceptives) and then applied to predict the PK and DDI of multiple doses of midostaurin (100 mg twice daily), with EES.
Search strategy used in Novartis Global Safety Database (Argus)To identify any post-marketing individual case report of potential DDI between concomitant hormonal contraceptives and midostaurin, a cumulative search until June 30, 2023, was conducted in the Novartis Global Safety Database (Argus) using the Medical Dictionary for Regulatory Activities (MedDRA; version 26.0) with SMQ (broad) pregnancy and neonatal topics. A second cumulative search was conducted until June 30, 2023, to retrieve all cases where the patient had taken midostaurin along with any of the oral contraceptives (estrogen, progestin, ethinylestradiol norgestrel or a combination of estrogen/progestin or ethinylestradiol/norgestrel) as concomitant/co-suspected medications.
Bioanalytical methodFor Study 1 with CYP substrates, plasma concentrations of pioglitazone and hydroxy-pioglitazone was assessed using a validated LC–MS/MS method with a LLOQ of 0.500 ng/mL and 500 ng/mL, respectively (PRA Health Sciences Assen Netherlands). Concentrations of midazolam and 1ʹ˗hydroxy midazolam in plasma were measured using a validated LC–MS/MS method with a LLOQ of 0.100 ng/mL for midazolam and 100 ng/mL for 1ʹ˗hydroxy midazolam (PRA). Concentrations of bupropion and 1ʹ˗hydroxy bupropion in plasma samples was quantified by a validated LC˗MS/MS method with a LLOQ of 0.5000 ng/mL and 250 ng/mL, respectively (PRA).
For Study 2 with oral contraceptives, concentrations of EES and LVG were measured in plasma with a LLOQ of 5.00 pg/mL and 500 pg/mL, respectively (SGS France).
For both Studies 1 and 2, the concentrations of midostaurin and its metabolites CGP52421 and CGP62221 in plasma were determined using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) assay with a lower limit of quantitation (LLOQ) of 10 ng/mL (SGS France).
留言 (0)