
記住我





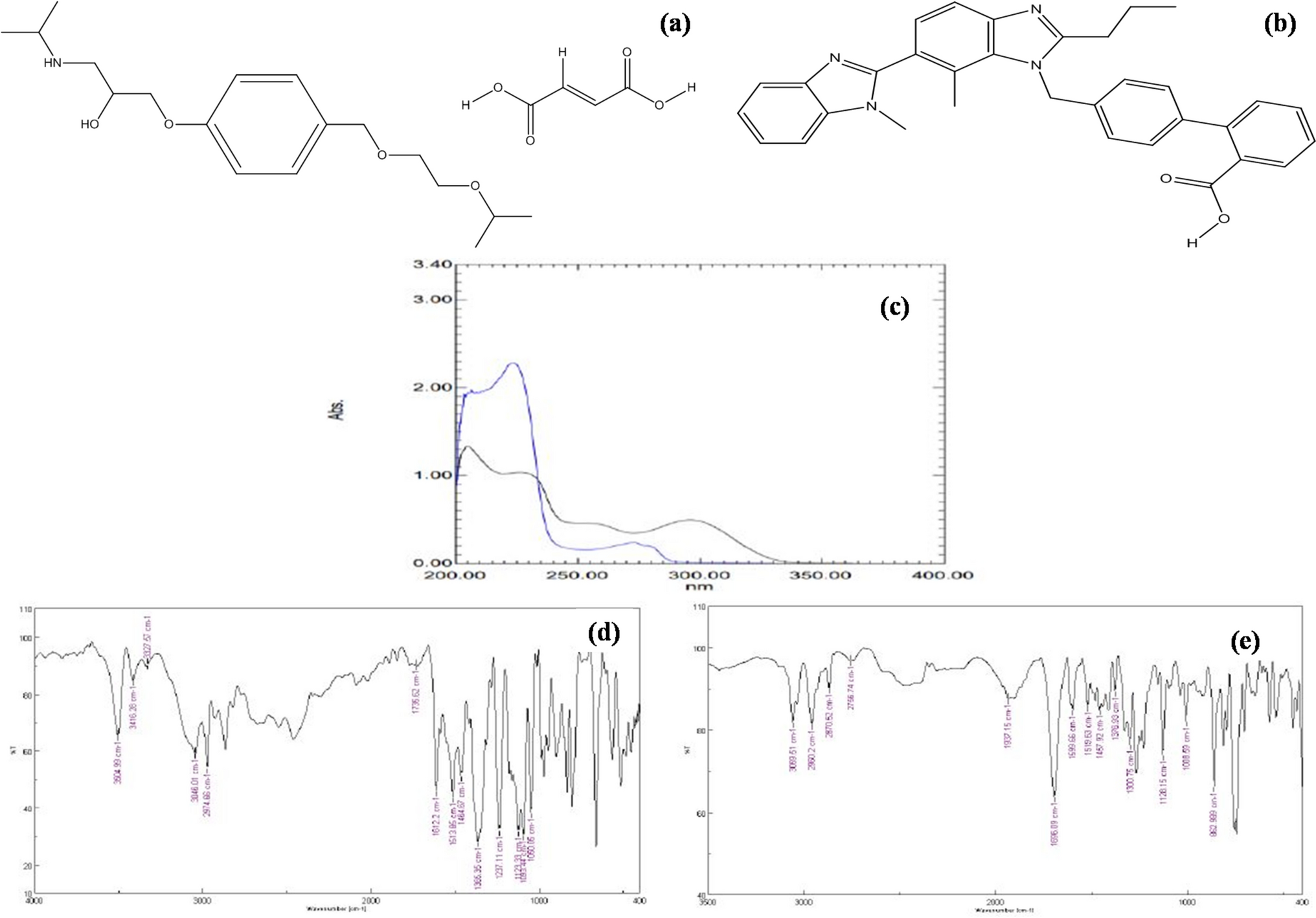



The UV-absorption spectra of the three medications under investigation show that for EMG, PGT, and RSV, their maximal absorbance wavelengths were at 230, 232, and 240 nm, respectively. After the substances under investigation had been completely separated on TLC plates, it was discovered that the wavelength 230 nm serves as an appropriate wavelength for the detection of the selected drugs as shown in Fig. 2.
Fig. 2
Absorption spectra of EMG, PGT, and RSV
3.2 Method optimization3.2.1 Mobile phase and saturation time effectsTrials for the selection of the mobile phase composition were based on the difference in polarity between EMG, PGT, and RSV. First, a mobile phase composed of 1,4-dioxane‒phosphate buffer (pH 4) was used with composition (5.0:5.0, V/V); however, 1,4-dioxane was a nonpolar solvent and therefore PGT split off to the opposite side of the polar silica gel plate while EMG and RSV traveled fast along the solvent front zone. After changing the ratio of the previous mobile phase to be of 3.0:7.0 (V/V) composition, no separation occurred. Another mobile phase system based on the utilization of ethyl acetate‒methanol was used with composition of 5.0:0.1 (V/V); however, no separation occurred. By adding toluene (nonpolar solvent) to the previous system, the system became toluene‒ethyl acetate‒methanol (4.0:4.0:2.0, V/V); EMG and RSV both had the same RF but PGT had a RF far from them, and by replacing toluene with n-hexane, as it is more nonpolar solvent than toluene, the three mentioned drugs were separated well from each other, but the RSV spot was very broad and so, by adding a modifier (glacial acetic acid) with a very small quantity (0.05 µL), the separation occurred very well and the broadness disappeared. The trials’ outcomes are reported in Table 1. So, n-hexane‒ethyl acetate‒methanol‒acetic acid (4.2:4:1.75:0.05, V/V) was found to be the ideal mobile phase composition that provided excellent RF values, acceptable peak shapes, and satisfactory resolution. Figure 3A depicts the 2D densitogram produced by this mobile phase system.
Table 1 The impact of utilizing various mobile phases for separating EMG, PGT, and RSV using the developed HPTLC methodFig. 3
A Two-dimensional and B three-dimensional HPTLC densitograms of a mixture containing EMG, PGT, and RSV (each 100 ng/band) at 230 nm using n-hexane‒ethyl acetate‒methanol‒glacial acetic acid (4.2:4:1.75:0.05, V/V)
To determine the ideal saturation period, experiments were conducted with various saturation times ranging from 10 to 30 min. The ideal saturation period was determined to be 20 min. The best saturation time was chosen based on the shortest amount of time that produced the best separation between two bands and the most reliable findings for RF.
3.3 Method validationIn accordance with the International Council for Harmonisation (ICH) criteria for the validation of analytical procedures, the suggested method was validated [49].
3.3.1 Calibration curveThe linearity of the method was evaluated by two tests: the lack-of-fit test and Mandel’s fitting test [50] (Table 2). The lack-of-fit test is a statistical method that checks how well a linear regression model fits the data. The lack-of-fit test showed that the linear regression model was good for all drugs. Mandel’s fitting test is another statistical method that measures the linearity of the calibration curve. The Mandel’s fitting test proved that the linear regression model worked well for the drugs EMG and PGT, but not for the drug RSV. The quadratic regression model had very slightly higher R2 and lower standard error values than the linear regression model for two drugs [50]. This means that both the linear and quadratic regression models fit the data of these drugs very well. Table 3 shows the comparison between the linear and quadratic regression parameters for the three drugs.
Table 2 Summary of the linear regression and validation findings in pure form of the investigated drugs simultaneouslyTable 3 Comparison between the linear and quadratic equation of the studied drugsLinear regression is a simpler, more robust, and more versatile model than quadratic regression. It is easier to use, faster to compute, and more resistant to overfitting. It can also handle outliers better. Quadratic regression should only be used if there is a strong theoretical or empirical basis to believe that the relationship between the independent and dependent variables is quadratic [50].
3.3.2 Linearity, quantification, and detection limitsPeak areas were created following simultaneous analysis of the standard drugs (EMG, PGT, and RSV synthetic mixture) using the suggested approach under ideal chromatographic conditions, where good linearities (5‒250 ng per band) for each drug with correlation coefficient values ranging from 0.9990 to 0.9992 were obtained. According to statistical estimates, the formulae for limit of detection (LOD) = 3.3 × standard deviation (SD) of intercept/slope and limit of quantitation (LOQ) = 10 × SD of intercept/slope (SD is the standard deviation). LOD and LOQ values (1.72‒5.23, 1.79‒5.42, and 1.52‒4.61 ng per band) were provided for the standard drugs: EMG, PGT, and RSV, respectively. The three-dimensional chromatograms and linearity plots are displayed in Fig. 3B. Table 2 displays the significant quantitative parameters; the statistical values show that the approach has good sensitivity for identifying the medications under investigation.
3.3.3 AccuracyThe closeness of agreement between the values regarded as a conventional true value or an approved reference value and the value found expresses the accuracy of an analytical method. As shown in Table 4, good recovery values and low percent relative standard deviation (%RSD) values demonstrated the suggested approaches’ accuracy. Six duplicates of each of three distinct concentrations; 50, 150, and 250 ng per band for EMG, PGT, and RSV were used to examine the accuracy of the proposed method.
Table 4 Accuracy and precision of the proposed approach3.3.4 PrecisionIntra-day and inter-day precision studies for EMG, PGT, and RSV were used to demonstrate the precision of the suggested analytical procedure. Six duplicates of each of three distinct concentrations: 50, 150, and 250 ng per band for each of the mentioned drugs were used to perform intra-day precision. For 3 days in a row, the same three concentrations, 50, 150, and 250 ng per band were used to measure inter-day precision. To identify any intra-day and inter-day variability, the %RSD value was calculated as shown in Table 4. The results showed that the %RSD did not exceed 3%, confirming the great precision of the suggested approach.
3.3.5 Selectivity and specificityThe analytical method’s selectivity refers to its capacity to identify analytes without interference from other matrix elements. The method’s selectivity was investigated by analyzing EMG, PGT, and RSV mixture spiked with human plasma sample on the TLC plate as seen in Fig. 4A and B showing that there were no interfering plasma component peaks and that the RF values of the examined medicines in human plasma were identical to those in the reference combination. The combined spectra of their individual tablet extracts, along with the superimposed spectra of the EMG, PGT, and RSV standard mixtures, confirm the suggested method’s high specificity and strong correlation. Figure 5 displays spectral comparison between tablets and standard mixture solutions, proving the purity and identity of the peaks.
Fig. 4
Two-dimensional HPTLC densitogram of A human plasma sample spiked with EMG, PGT, and RSV (each 100 ng per band), B blank plasma sample, C real rat plasma following administration of single oral dose of EMG (10 mg/kg), PGT (3 mg/kg), and RSV (200 mg/kg) after their reported Tmax, and D blank rat plasma
Fig. 5
Spectra comparison of A standard solution containing 150 ng per band of (EMG) and tablet extract sample solution 150 ng per band of EMG, B standard solution containing 150 ng per band of (PGT) and tablet extract sample solution 150 ng per band of PGT, and C standard solution containing 150 ng per band of (RSV) and tablet extract sample solution 150 ng per band of RSV
3.3.6 RobustnessRobustness is a measure of method capacity to persist unaltered after minor but intended variations in the parameters of an analytical process. Saturation duration, development distance and ratios of the mobile phase (n-hexane‒ethyl acetate‒methanol‒glacial acetic acid) underwent minor alterations that were evaluated. The robustness of the approach illustrated in Table 5 showed good recoveries and low %RSD values.
Table 5 Robustness findings of the proposed TLC method for determining EMG, PGT, and RSV, simultaneously3.4 Applications of the suggested approach3.4.1 Pharmaceutical formulationsThe proposed HPTLC method was successfully used to identify EMG, PGT, and RSV in the locally produced tablets. The average recovery rates ± RSD for the manufactured tablets for EMG, PGT, and RSV, respectively, were (99.66 ± 1.10), (100.93 ± 0.34), and (101.41 ± 1.71), according to the procedure described in the study. The results of the proposed methods and the outcomes of reported methods [51,52,53] were compared statistically. The simultaneous study of EMG, PGT, and RSV in their dose forms utilizing the suggested protocol, as given in Table 6, was carried out with good accuracy and precision because no significant differences were detected using applications of t test and F test at 95% confidence level.
Table 6 Using the proposed method and the reported method, EMP, PGZ ,and ROS dose forms were analyzed (n = 6) [51,52,53]3.4.2 Spiked human plasmaThree medications, EMG, PGT, and RSV, can be detected simultaneously in spiked human plasma using the solid-phase extraction (SPE). Advantages of SPE are low intrinsic costs, little solvent usage, and being a less complex processing method [54]. Calibration curves for the three investigated drugs spiked with human plasma were established. A linear relationship was found between the integrated peak area and the concentrations of EMG, PGT, and RSV spiked with human plasma over the concentration range of 5‒250 ng per band. The LOD and LOQ results were found to be (1.29‒3.93), (1.68‒5.10), and (1.12‒3.38) ng per band for EMG, PGT, and RSV, respectively. Table 7 lists all regression analysis information obtained from spiked human plasma. After examining human plasma samples at three different concentration levels, good recovery percentages and low %RSD values were discovered (Table 8). By showing a high level of drug extraction efficiency from plasma without interference from intrinsic plasma constituents, these results illustrated the selectivity of the method (Fig. 4A and 4B). The results showed that the suggested procedure may be used on real plasma.
Table 7 An overview of the results of linear regression in spiked human plasma and recovery of mentioned drugs in real rat plasmaTable 8 Results of the proposed method in spiked human plasma3.4.3 Real rat plasmaBy measuring the amount of EMG, PGT, and RSV simultaneously in real rat plasma, the effectiveness of the suggested approach was further assessed. After giving a single oral dose of EMG 10 mg/kg, PGT 3 mg/kg, and RSV 200 mg/kg to five male Wistar rats, blood samples were taken. From reported studies of the EMG, PGT, and RSV in rat plasma, EMG (3 mg/kg by oral route) was metabolized by glucuronidation, its Cmax was 0.167 ± 0.829 µg/mL after Tmax 2 h and excreted in feces mainly. PGT (3 mg/kg by oral route) was metabolized by hydroxylation and oxidation via liver, its Cmax was 3.87 ± 0.15 µg/mL after Tmax 2.67 ± 0.52 h and approximately 15‒30% of a PGT dose was removed in the urine as metabolites, with the remainder excreted either unaltered or as metabolites in the bile. RSV (200 mg/kg by oral route) had N-desmethylrosuvastatin, a less potent primary metabolite, its Cmax was 3.04 µg/mL after Tmax 0.87 ± 0.29 h and excreted in feces mainly (90%) [46,47,48]. It was found that the resolution and peak shape of the real samples were comparable to those obtained when utilizing the reference samples, and there were no interference peaks seen (Figs. 4C and 4D). The recovery percentage obtained by dividing the response of samples by that of standards using the same concentration level for both sample and standard was in the range of 90.97 ± 3.13 to 94.64 ± 2.12 (Table 7).
3.5 Greenness evaluation of the developed HPTLC technique and comparison with the previously reported methodsTo assess the environmental impact of analytical procedures, additional metrics have been developed in recent years. To compare and evaluate various analytical methods more effectively in terms of their green characteristics, it is often advised to use a variety of color evaluation tools. Three new techniques have been established to evaluate greenness in our research: the analytical eco-scale [55], the green analytical procedure index (GAPI) [56], and the new analytical greenness metric (AGREE) [57].
The analytical eco-scale [55] has the advantage of being semi-quantitative and having a straightforward scoring system. The eco-scale score is determined by deducting from 100 the number of penalty points awarded to each component (reagent amount and nature, occupational hazard, energy consumption, and amount of created waste) that does not adhere to the ideal green analysis standards. The scores above 75 indicate a very good quality green analysis, the values between 50 and 75 indicate a good green analysis, and the scores below 50 indicate a poor green analysis. The developed methods received an analytical eco-scale score of 76, which makes it a very good green analysis with no adverse environmental impact (Table 9).
Table 9 Eco-scale penalty points for the developed HPTLC method for determining EMG, PGT, and RSV simultaneouslyThe new GAPI index [56] has the advantage of covering the entire analytical process when compared with the analytical eco-scale [55]. There are five pentagrams, each representing a step in the analytical process, such as collecting the samples, preparing the samples, using reactive and solvents in the apparatus, and the objective of the analytical process. GAPI has three color coding systems: red denotes a high environmental risk, whereas yellow and green denote a lower risk and greater greenness. Three other previously mentioned analytical separation methods [17,18,19] have been compared with the newly developed HPTLC method. Table 10 displays the GAPI index for the suggested method evaluation along with three published analytical techniques for the examination of the three substances under investigation. All of these methods can identify two of the studied medications; however, our method can identify all three of the studied medications simultaneously. According to Table 10a, the suggested green HTPLC method’s GAPI index has nine green, four yellow, and two red pentagrams. The methods shown in Table 10b‒d at this time have three red pentagrams. The red areas show a big ecological impact, the yellow areas show a smaller ecological impact, and the green areas show a greater ecological impact.
Table 10 Comparison of the developed HPTLC method with some reported separation methodsAn additional assessment of greenness was made utilizing the innovative, more inclusive, and informative AGREE tool [57] to produce the most accurate and logical rating possible. The 12 green analytical chemistry (GAC) principles are taken into account by this tool, which is an advantage. As a result, a thorough assessment of each method’s green color was made, and a thorough distinction between the developed and reported methods’ green characteristics was established. The AGREE tool demonstrated the superiority of our created HPTLC method, which received the best analytical score (0.81) from AGREE while using only 1 mL per sample of waste.
AGREE scores and waste amounts of the mentioned reported methods were 0.67 and 9 mL, 0.66 and 2 mL, and 0.58 and 11.8 mL [17,18,19] for the reported methods presented in Table 10b‒d. All of the pictograms for the developed and reported methods are included in Table 10. Finally, after comparing the three different methods, we found that our method HPTLC is more environmentally friendly than the other methods mentioned.
Furthermore, our developed method is the most sensitive method and used to detect the quantities of the drugs mentioned earlier in spiked human plasma and real rat plasma as the mentioned reported methods have very low sensitivity and all applied in dosage form only.
留言 (0)