Patients
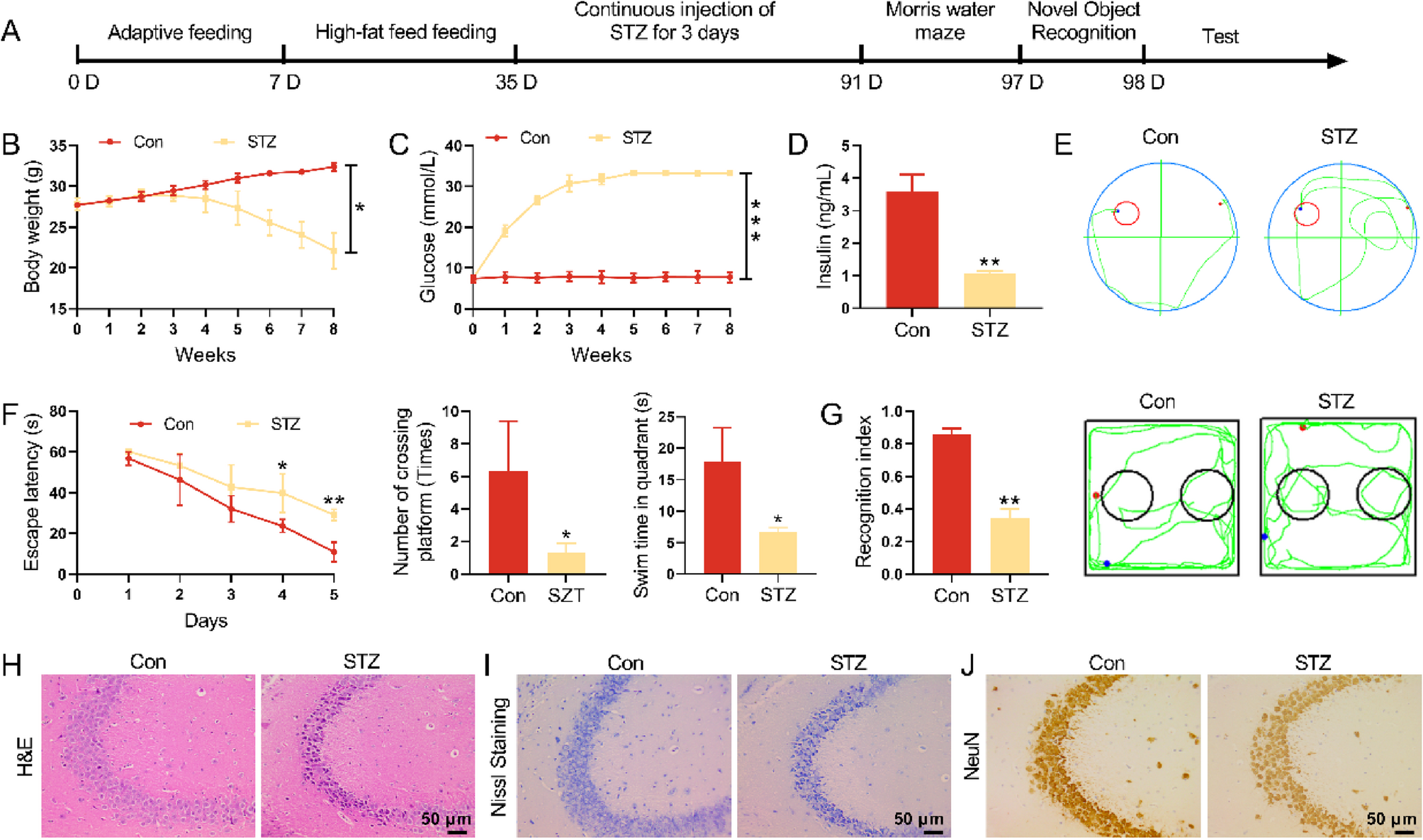
The Ethics Committee of the Second Affiliated Hospital of the Air Force Medical University of China approved this study (the registered number for this retrospective study is 202101–08). From September 2021 to March 2022, 77 emergency room patients aged 56.7 \(\pm\) 10.9 years who suffered ICH with hematomas of 15.13 \(\pm\) 2.7 ml were enrolled in this study. Patients with severe neurological or hepatic diseases or other severe systemic diseases were excluded. The Glasgow Coma Scale (GCS) was used to evaluate the severity of coma (Teasdale and Jennett 1974). The serum level of FGF21 at 24 h from the onset of ICH was measured. Patients’ GCS scores on hospital admission and at discharge were also recorded. \(\Delta\)GCS% indicates the following ratio: (GCSdischarge − GCSadmission)/GCSadmission. Positive values indicate neurological improvement, while negative values indicate deterioration during hospitalization. According to the \(\Delta\)GCS%, patients were assigned to two groups: the \(\Delta\)GCS% > 0 group and the \(\Delta\)GCS% < 0 group. The relationship between the serum level of FGF21 and the \(\Delta\)GCS% was determined. The detailed experimental protocols are listed in Additional file 1.
Animals
A committee of the Second Affiliated Hospital of the Air Force Medical University of China reviewed and approved the protocols for these experiments, and these experiments were performed strictly according to the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6 mice (25–30 g) were purchased from the Animal Center of the Air Force Medical University. They were divided into 4 groups to determine the protective effects of FGF21 on the neural system and BBB in vivo (the detailed experimental protocols are provided in Additional file 1). SIRT6flox/flox homozygous mice were purchased from the Shanghai Model Organisms Center (NM-CKO-200241). Through the crossbreeding of SIRT6flox/flox mice with mice expressing Cre recombinase under the control of the vascular endothelial-specific cadherin promoter (Cdh5-CreERT2,NM-KI-200173, ShangHai model organisms), we generated Cdh5-CreERT2-SIR6flox/flox mice. Next, according to the manufacturer’s instructions (T5648, Sigma‒Aldrich), we dissolved tamoxifen in corn oil and then made a mixture of tamoxifen and corn oil at a concentration of 20 mg/ml. This mixture was injected intraperitoneally (20 mg/kg) every 24 h for 5 consecutive days to obtain eSIRT6−/− mice. Meanwhile, SIRT6flox/flox homozygous littermates that did not receive tamoxifen injection were utilized as controls to assess the involvement of the SIRT6 pathway (the detailed experimental protocols are provided in Additional file 1). Recombinant FGF21 (C600252) was purchased from Sangon Biotech and intraperitoneally administered (1.5 mg/kg) immediately after the induction of ICH for 7 days. All mice were kept in special facilities with proper humidity and temperature and were provided with abundant food and water.
Induction of ICH in vivo
The mice were anesthetized with 1% pentobarbital (50 ml/kg) and fixed to a stereotactic frame. An approximately 0.5 cm longitudinal incision was made along the midline of the head to expose the bregma. The coordinates of the drill hole relative to bregma were as follows: 2.0 mm lateral and 0.4 mm anterior. A microsyringe was used to vertically penetrate 3 mm into the cortex, and approximately 0.5 µl of 0.05% collagenase (C5138, Sigma) was injected into the striatum. The microsyringe was withdrawn after 6 min, and the wound was sealed. The mice in the sham group experienced all operative processes except for the injection of 0.5 µl 0.05% collagenase, which was the key difference with the ICH, ICH + Vehicle and ICH + FGF21 groups. For the ICH + Vehicle group, normal saline was employed to simulate the process of intraperitoneal injection of FGF21 in the ICH + FGF21 group. Areas of interest were selected from perihematoma spots 1.5 mm away from the hematoma (Fang et al. 2019).
Behavioral assessments
The modified Neurological Severity Score (MNSS) was utilized to assess sensorimotor deficits in mice before and after the induction of ICH. The MNSS is capable of evaluating sensory and motor deficits through an elaborate grading system (score of 0–18) (Shi et al. 2021; Liu et al. 2023). A higher score indicates more severe damage. The tests were performed by two blinded observers. The detailed rating scale is listed in Additional file 3.
A 30° device was employed to perform the corner turn test (Shi et al. 2021). Mice were allowed to proceed toward the device and freely choose the direction of turning at the corner before and after ICH. The number of right-turn choices was recorded during 10 repeat tests by 2 blinded observers.
For the wire hanging test (Shi et al. 2021; Zhu et al. 2014), the hind limbs of each mouse were tied, and the mouse was placed on a metallic wire stretched between two posts approximately 40 cm above the ground. A soft pillow was placed under the wire to avoid injury caused by a fall. Before and after ICH, the mice were allowed to grasp the wire tightly, and the duration before falling was recorded.
Measurement of brain water content
Seventy-two hours after ICH, the brains of the mice were removed and weighed. The data were recorded as the wet weight. Next, we put the brains into an oven for 72 h at temperatures from 95 to 100 °C. These brains were then weighed again, and the data were recorded as the dry weight. Brain water content was assessed and calculated using the following formula: (wet weight-dry weight)/wet weight × 100%.
Magnetic resonance imaging (MRI)
After anesthetization, the mice were fixed to a special device to fix their head and restrict movement. With a Discovery MR750 3.0T scanner (General Electric Company, USA), the lesion volume of each mouse was measured 72 h after onset. T2-weighted pictures were selected. Fiji software was employed to assess and analyze the results.
Evans blue staining
Mice were fixed in a special device with their tails anchored at an illuminated groove. After massaging the tail and temporarily blocking the blood flow, the tail vein could be easily spotted. Two hundred microliters of 2% Evans blue was injected with a microsyringe. Seventy-two hours later, the mice were sacrificed, and their brains were sliced for the following procedures. Evans blue extravasation was measured and analyzed according to a previous study (Radu and Chernoff 2013).
Transmission electron microscopy
Mice were sacrificed and perfused with 0.9% saline and 4% paraformaldehyde at 72 h after ICH. The brains were removed and coronally sliced into 2 mm slices. Then, slices that contained a hematoma were selected and immersed in 4% glutaraldehyde overnight. We used 1% osmium tetroxide to fix these slices for 1 h. Next, the slices were dehydrated with ethanol and embedded in resin. Finally, the slices were cut into 80-nm sections and observed with a JEM-1400 electron microscope (JEOL, Tokyo, Japan).
Cultured human brain microvascular endothelial cells (HCMECs)
Human brain microvascular endothelial cells (HCMECs) were purchased from iCell Corporation (iCell-h070, China) and cultivated in EBM-2 supplemented with penicillin‒streptomycin in cell culture plates. SIRT6 silencing was performed with a small interfering RNA targeting SIRT6 (siSIRT6) in vitro. Invalid small interfering RNA was used as a negative control (si-NC). Lipofectamine™ 2000 was employed for siRNA transfection according to the manufacturer’s instructions (18324012-012, Invitrogen, USA) after the cells reached a proper density. HCMECs were randomly pretreated with FGF21/vehicle/siSIRT6/si-NC before exposure to oxyhemoglobin. (the detailed experimental protocols are described in Additional file 1, siSIRT6: 5ʹ-CAAGUGUAAGACGCAGUACGUTT-3ʹ forward and 5ʹ-ACGUACUGCGUCUUACACUUGTT-3ʹ reverse, si-NC: 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ forward and 5ʹ-ACG UGACACGUUCGGAGAATT-3ʹ reverse). After incubation with oxyhemoglobin for 2 h, the cell samples were collected for subsequent procedures.
Cell viability test
Cell viability was measured using a CCK-8 kit (96992, Sigma). Approximately 10 µl of HCMECs per well were seeded in a 96-well plate according to the manufacturer’s instructions. After 24 h, the cells were assigned to different groups to receive different treatments according to the experimental protocols. Then, approximately 10 µl of CCK-8 solution was added to each well. Four hours later, a microplate reader was used to measure the absorbance at a wavelength of 450 nm.
Permeability of endothelial cells
HCMECs were seeded into 24-well transwell chambers (1 × 105 per well) with 200 µl of EBM-2 for 48 h (Adil and Somanath 2021). When cells reached a compact density, FGF21/vehicle/siSIRT6/si-NC was added to the chambers. Then, oxyhemoglobin was added to the related chambers for 2 h. After that, the medium was replaced with medium containing 1% FITC-dextran (10 mg/ml, Sigma). Three hours later, the medium was withdrawn, and the fluorescence of each well was measured at 485 nm excitation and 520 nm emission wavelengths with an EnSpire multimode microplate reader with EnSpire Manager software (PerkinElmer Company, USA). A higher fluorescence signal indicated more damage to the integrity of HCMEC monolayers.
ELISA
Patient whole-blood samples were collected in EDTA-treated tubes and centrifuged at 12,000×g for 15 min to obtain the plasma. Then, with a specific ELISA kit (NeoBioscience, China), the level of FGF21 was measured. All procedures were strictly performed according to the manufacturer’s instructions.
Western blotting
Samples of proteins were separated by SDS‒PAGE and transferred to PVDF membranes. The membranes were then blocked with 5% nonfat milk for 1 h. Next, specific primary antibodies were added to the box containing the blocked membranes, and the membranes were incubated overnight. After rinsing twice, the membranes were incubated with the appropriate corresponding horseradish peroxidase–conjugated anti-rabbit secondary antibodies (1:5000, AS014, ABclonal) for 2 h. Then, pictures of targeted proteins were captured with the assistance of a Bio-Rad Imaging System (Bio-Rad). ImageJ was employed to analyze the bands. The primary antibodies utilized in this experiment included anti-ZO-1 (1:1000, 7773-1-AP, Proteintech), anti-occludin (1:1000, 272601-1-AP, Proteintech), anti-SIRT6 (1:1000, 13572-1-AP, Proteintech), anti-mfn1 (1:1000, 14739, Cell Signaling), anti-mfn2 (1:1000, 11925, Cell Signaling), and anti-Drp1 (1:1000, 14647, Cell Signaling).
Immunofluorescence (IF) and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining
After dehydration, the mouse brains were cut into 30 µm slices and incubated with 1% Triton X-100. Then, the slices were incubated with primary antibodies such as anti-CD31 (1:500, AF806, R&D) and anti-SIRT6 (1:500, 13572-1-AP, Proteintech) overnight at 4 °C. The slices were incubated with species-specific secondary antibodies with different fluorescence signals for 2 h. The nuclei were labeled with DAPI (1:1000, Invitrogen). All slices were imaged with an A1 Si confocal microscope (Nikon) and analyzed by Fiji software. TUNEL staining was performed strictly according to the manufacturer’s instructions (C1086, Beyotime). Areas of interest were selected from perihematoma spots 1.5 mm away from the hematoma (Fang et al. 2019).
Cell samples were seeded on confocal plates. After cultivation to an appropriate density, paraformaldehyde was utilized to fix the samples. Then, the same protocols described above were utilized to treat the cell samples.
Measurement of mitochondrial morphology and function
HCMECs were cultured on a confocal plate. After the cells reached the appropriate density and were treated with different protocols, approximately 10 nM MitoTracker Red (M22426, Invitrogen) was added to the plate, and the cells were incubated for 30 min. Thereafter, with a fluorescence microscope (A1 Si, Nikon), the morphology of mitochondria was captured and then analyzed with Fiji software according to a previous study (Valente et al. 2017). Additionally, 10 nM MitoSOX Red (M36008, Thermo) was added to the plate to measure the levels of ROS in mitochondria, and the results were analyzed with Fiji software. In addition, HCMECs were incubated with 10 nM JC-1 (C2006, Beyotime) for 30 min to assess the depolarization of the membrane. Commercial assay kits were used to measure manganese superoxide dismutase (MnSOD) activity (JL20470, Jiang Lai). For ATP measurement, according to the manufacturer’s instructions (BC0300, Solarbio), samples were lysed and centrifuged. The supernatants were mixed with the ATP detection working solution, and the ATP levels of the different groups were measured with a standard curve.
Reverse transcription and quantitative real-time PCR
Real-time PCR was applied to assess gene expression in samples. Total RNA was extracted from HCMEC samples using TRI Reagent (Invitrogen Carlsbad, CA) according to the manufacturer’s protocols. Then, the cellular RNA was converted to cDNA in a final volume of 20 μl. All RT‒PCR experiments were performed using Master Mix provided by Thermo Fisher. Each reaction (20 μl) contained 2 μl cDNA, 400 fmol of each primer and 10 μl of Master Mix. The following primers were employed: for SIRT6, 5ʹ-GCT TCC TGG TCA GCC AGA-3ʹ (forward) and 5ʹ-CTT GGC ACA TTC TTC CAC AA-3ʹ (reverse); for β-actin, 5ʹ-GCA CAG AGC CTC GCC TT-3ʹ (forward) and 5ʹ-GTT GTC GAC GAC GAG CG-3ʹ (reverse). All qPCR assays were performed in triplicate in a 96-well plate according to the manufacturer’s protocol. The results were normalized against those of β-actin and expressed as fold changes for the relative mRNA expression levels.
Statistical analysis
SPSS Statistics 20.0 (IBM) was employed to analyze all data. The mean and standard deviation are used for normally distributed data. The median is used for nonnormally distributed data. Categorical data are presented as frequencies. Univariate and multivariate logistic regression models were used to explore independent risk factors for unfavorable outcomes. Multiple groups were compared using one-way analysis of variance (ANOVA) or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. Student’s t tests were used to compare 2 groups. Spearman correlation analysis was used to test the correlation between two quantitative variables. GraphPad Prism 6.0 software was employed for statistical analysis and drawing. Fiji software was used to quantify the targeted areas and fluorescence signals. Differences with P values < 0.05 were defined as statistically significant.



Comments (0)