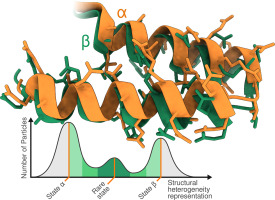
Proteins are the major components of the molecular machinery that keep cells and organisms alive, performing their tasks in a spatially and temporally organized fashion. Thus, their structures and dynamics have evolved to ensure optimal function [1,2]. Motions are a pivotal intrinsic property of proteins, exploited by nature to fine-tune their reactivities and interactions, when they perform their activities [3, 4, 5]. Alas, protein dynamics are generally ignored in the traditional representations of static average structures, despite their fundamental importance for understanding mechanisms and regulation of biological processes.
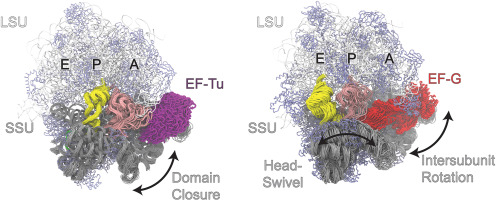
Protein motions span over 15 orders of magnitude in timescale, ranging from subpicoseconds to milliseconds or seconds [6]. Subpicosecond and picosecond dynamics are associated with vibrational motions of atoms, pico- to nanosecond motions are connected to side chain reorientations, while slower motions involve the collective movement of different parts of a protein, ranging from covalent bond isomerization (disulfides, cis-trans proline) to local or loop conformational changes to reorientation of domains [7]. Functionally relevant conformational rearrangements in proteins are often associated with changes in dynamics [8], both in frequencies and amplitudes. They can also occur in regions distal to the structural change by allosteric mechanisms [9,10].
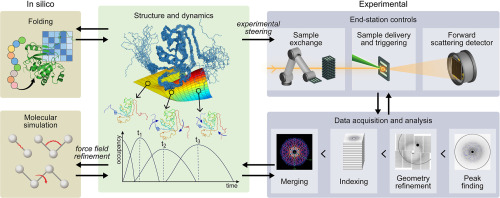
Among the multitude of current methods for atomic-level structure determination, nuclear magnetic resonance (NMR), cryogenic electron microscopy (cryo-EM) and X-ray crystallography provide the spatial arrangements of atoms in a protein. The resulting protein structural models can be complemented by computationally assessing dynamics using large-scale molecular dynamics (MD) simulations [11, 12, 13]. Dynamics can also be explored directly by NMR: timescales from pico- to milliseconds can be examined through relaxation measurements, lineshape analysis, and exchange experiments [14,15]. By contrast, in cryo-EM and X-ray crystallography, information on timescales of motions is absent, although data on conformational distributions present in the density maps are often interpreted as reflecting possible motions [16, 17, 18]. Timescales of motions accessible by MD simulations critically depend on the length of simulations [19] and usually do not exceed milliseconds for all-atom simulations on large systems [20, 21, 22, 23]. Furthermore, many large-amplitude conformational changes are not accessible by MD simulations, and time-independent approaches, such as normal mode analysis, principal component analysis or time-structure independent component analysis, are necessary [24, 25, 26]. Gaining information about the entire range of biologically relevant timescales of protein motions requires integration of all possible experimental and computational techniques to visualize dynamics and connect motions to function.
In this review, we will highlight recent impactful studies of protein dynamics using an integrative approach encompassing NMR, cryo-EM, and MD simulations, reporting on functionally significant motions in a number of complex biological systems, including enzymes and large molecular assemblies. Such integrative studies are still relatively rare, and in the second part of the review investigations integrating NMR and cryo-EM will be highlighted.







留言 (0)